Es ist seit langem bekannt, dass jeder Mensch anders auf Umweltchemikalien reagiert. Die jüngste Explosion in der Molekularbiologie und Genetik hat zu einem klareren Verständnis der molekularen Grundlage einer solchen Variabilität geführt. Zu den wichtigsten Determinanten der individuellen Reaktion auf Chemikalien gehören wichtige Unterschiede zwischen mehr als einem Dutzend Superfamilien von Enzymen, die zusammenfassend bezeichnet werden xenobiotisch- (körperfremd) bzw Droge-metabolisierend Enzyme. Obwohl die Rolle dieser Enzyme klassischerweise als Entgiftung angesehen wird, wandeln dieselben Enzyme auch eine Reihe von inerten Verbindungen in hochgiftige Zwischenprodukte um. Kürzlich wurden viele subtile sowie grobe Unterschiede in den diese Enzyme kodierenden Genen identifiziert, die nachweislich zu deutlichen Variationen in der Enzymaktivität führen. Es ist nun klar, dass jedes Individuum eine unterschiedliche Ergänzung von Xenobiotika-metabolisierenden Enzymaktivitäten besitzt; Diese Vielfalt könnte als „metabolischer Fingerabdruck“ betrachtet werden. Es ist das komplexe Zusammenspiel dieser vielen verschiedenen Enzym-Superfamilien, das letztendlich nicht nur das Schicksal und das Toxizitätspotenzial einer Chemikalie bei einem bestimmten Individuum bestimmt, sondern auch die Bewertung der Exposition. In diesem Artikel haben wir uns entschieden, die Superfamilie der Cytochrom-P450-Enzyme zu verwenden, um die bemerkenswerten Fortschritte zu veranschaulichen, die beim Verständnis der individuellen Reaktion auf Chemikalien erzielt wurden. Die Entwicklung relativ einfacher DNA-basierter Tests zur Identifizierung spezifischer Genveränderungen in diesen Enzymen liefert nun genauere Vorhersagen der individuellen Reaktion auf Chemikalienexposition. Wir hoffen, dass das Ergebnis eine präventive Toxikologie sein wird. Mit anderen Worten, jeder Einzelne könnte etwas über die Chemikalien erfahren, auf die er oder sie besonders empfindlich reagiert, und so eine zuvor unvorhersehbare Toxizität oder Krebs vermeiden.
Obwohl dies im Allgemeinen nicht anerkannt wird, sind Menschen täglich einem Sperrfeuer unzähliger verschiedener Chemikalien ausgesetzt. Viele dieser Chemikalien sind hochgiftig und stammen aus einer Vielzahl von Umwelt- und Nahrungsquellen. Die Beziehung zwischen solchen Expositionen und der menschlichen Gesundheit war und ist ein Hauptaugenmerk biomedizinischer Forschungsbemühungen weltweit.
Was sind einige Beispiele für dieses chemische Bombardement? Mehr als 400 Chemikalien aus Rotwein wurden isoliert und charakterisiert. Mindestens 1,000 Chemikalien werden schätzungsweise durch eine angezündete Zigarette produziert. Es gibt unzählige Chemikalien in Kosmetika und parfümierten Seifen. Eine weitere wichtige Quelle der Exposition gegenüber Chemikalien ist die Landwirtschaft: Allein in den Vereinigten Staaten werden Ackerland jedes Jahr mehr als 75,000 Chemikalien in Form von Pestiziden, Herbiziden und Düngemitteln ausgesetzt; Nach der Aufnahme durch Pflanzen und Weidetiere sowie Fische in nahe gelegenen Gewässern nimmt der Mensch (am Ende der Nahrungskette) diese Chemikalien auf. Zwei weitere Quellen für große Konzentrationen von Chemikalien, die in den Körper aufgenommen werden, sind (a) chronisch eingenommene Medikamente und (b) die Exposition gegenüber gefährlichen Stoffen am Arbeitsplatz über ein ganzes Berufsleben hinweg.
Es ist inzwischen allgemein bekannt, dass die Exposition gegenüber Chemikalien viele Aspekte der menschlichen Gesundheit beeinträchtigen und chronische Krankheiten und die Entwicklung vieler Krebsarten verursachen kann. In den letzten zehn Jahren begann man, die molekulare Grundlage vieler dieser Beziehungen zu enträtseln. Darüber hinaus hat sich die Erkenntnis herauskristallisiert, dass sich Menschen in ihrer Anfälligkeit für die schädlichen Wirkungen einer Exposition gegenüber Chemikalien deutlich unterscheiden.
Gegenwärtige Bemühungen, die Reaktion des Menschen auf eine Exposition gegenüber Chemikalien vorherzusagen, kombinieren zwei grundlegende Ansätze (Abbildung 1): die Überwachung des Ausmaßes der Exposition des Menschen durch biologische Marker (Biomarker) und die Vorhersage der wahrscheinlichen Reaktion einer Person auf ein bestimmtes Expositionsniveau. Obwohl diese beiden Ansätze äußerst wichtig sind, sollte betont werden, dass sich die beiden deutlich voneinander unterscheiden. Dieser Artikel konzentriert sich auf die Genetische Faktoren zugrunde liegende individuelle Anfälligkeit für eine bestimmte chemische Belastung. Dieses Forschungsfeld wird weit gefasst Ökogenetik, oder Pharmakogenetik (siehe Kalow 1962 und 1992). Viele der jüngsten Fortschritte bei der Bestimmung der individuellen Anfälligkeit für chemische Toxizität sind aus einer größeren Wertschätzung der Prozesse entstanden, durch die Menschen und andere Säugetiere Chemikalien entgiften, und der bemerkenswerten Komplexität der beteiligten Enzymsysteme.
Abbildung 1. Die Wechselbeziehungen zwischen Expositionsbeurteilung, ethnischen Unterschieden, Alter, Ernährung, Ernährung und genetischer Anfälligkeitsbeurteilung – die alle beim individuellen Toxizitäts- und Krebsrisiko eine Rolle spielen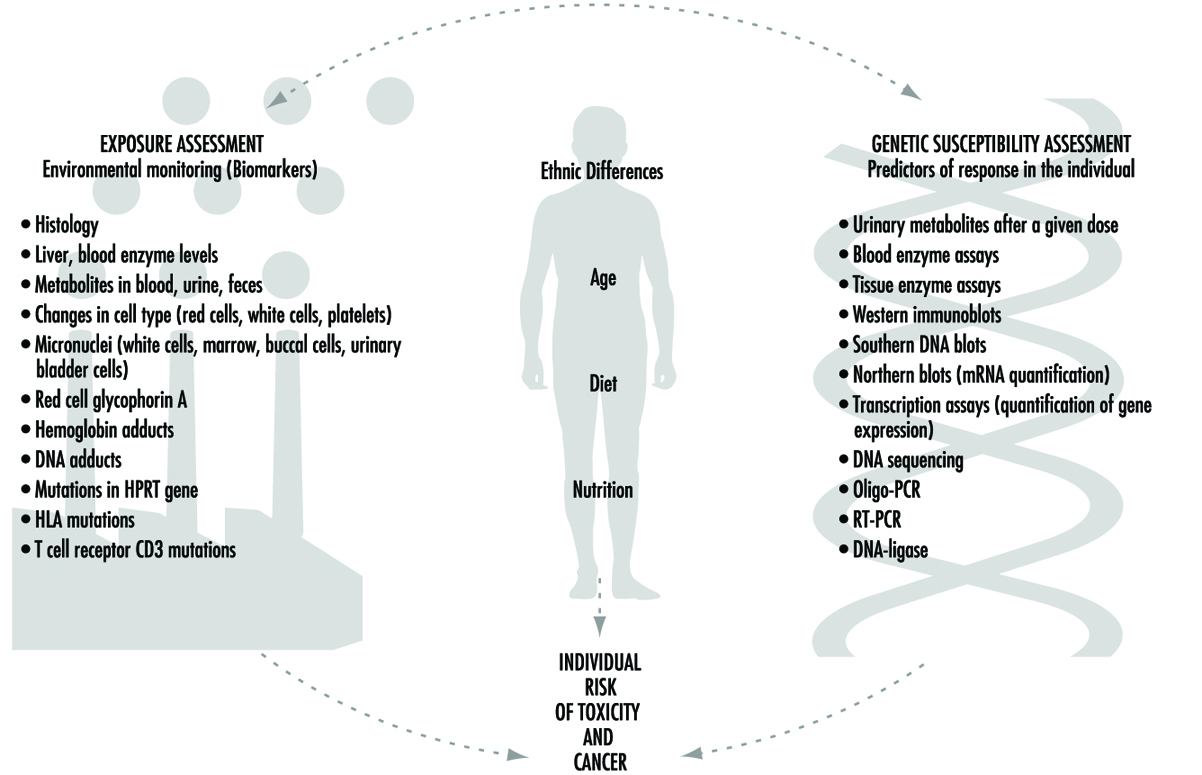
Wir werden zunächst die Variabilität toxischer Reaktionen beim Menschen beschreiben. Wir werden dann einige der Enzyme vorstellen, die für eine solche Variation der Reaktion aufgrund von Unterschieden im Metabolismus fremder Chemikalien verantwortlich sind. Als nächstes werden die Geschichte und Nomenklatur der Cytochrom-P450-Superfamilie detailliert beschrieben. Fünf menschliche P450-Polymorphismen sowie mehrere Nicht-P450-Polymorphismen werden kurz beschrieben; diese sind für menschliche Unterschiede in der toxischen Reaktion verantwortlich. Wir werden dann ein Beispiel diskutieren, um den Punkt zu betonen, dass genetische Unterschiede bei Individuen die Expositionsbewertung beeinflussen können, wie durch Umweltüberwachung bestimmt. Abschließend diskutieren wir die Rolle dieser Xenobiotika-metabolisierenden Enzyme in kritischen Lebensfunktionen.
Variation der toxischen Reaktion in der menschlichen Bevölkerung
Toxikologen und Pharmakologen sprechen gemeinhin von der durchschnittlichen tödlichen Dosis für 50 % der Bevölkerung (LD50), die durchschnittliche maximal tolerierte Dosis für 50 % der Bevölkerung (MTD50) und die durchschnittliche wirksame Dosis eines bestimmten Medikaments für 50 % der Bevölkerung (ED50). Wie wirken sich diese Dosen jedoch auf jeden von uns individuell aus? Mit anderen Worten, ein hochsensibles Individuum kann 500-mal stärker betroffen oder 500-mal wahrscheinlicher betroffen sein als das resistenteste Individuum in einer Population; für diese Leute, die LD50 (und MTD50 und ED50) Werte hätten wenig Bedeutung. LD50, MTD50 und ED50 Werte sind nur relevant, wenn sie sich auf die Gesamtbevölkerung beziehen.
Figure 2 veranschaulicht eine hypothetische Dosis-Wirkungs-Beziehung für eine toxische Reaktion von Individuen in einer gegebenen Population. Dieses generische Diagramm könnte bronchogenes Karzinom als Reaktion auf die Anzahl der gerauchten Zigaretten, Chlorakne als Funktion des Dioxinspiegels am Arbeitsplatz, Asthma als Funktion der Luftkonzentration von Ozon oder Aldehyd, Sonnenbrand als Reaktion auf ultraviolettes Licht, verringerte Gerinnungszeit usw. darstellen eine Funktion der Einnahme von Aspirin oder Magen-Darm-Beschwerden als Reaktion auf die Anzahl der Jalapeno Paprika verzehrt. Im Allgemeinen gilt in jedem dieser Fälle, je größer die Exposition, desto größer die toxische Reaktion. Der Großteil der Bevölkerung zeigt den Mittelwert und die Standardabweichung der toxischen Reaktion als Funktion der Dosis. Der „resistente Ausreißer“ (unten rechts in Abbildung 2) ist eine Person, die bei höheren Dosen oder Expositionen weniger stark anspricht. Ein „empfindlicher Ausreißer“ (oben links) ist eine Person, die auf eine relativ geringe Dosis oder Exposition übertrieben reagiert. Diese Ausreißer mit extrem unterschiedlichen Reaktionen im Vergleich zur Mehrheit der Individuen in der Bevölkerung können wichtige genetische Varianten darstellen, die Wissenschaftlern dabei helfen können, die zugrunde liegenden molekularen Mechanismen einer toxischen Reaktion zu verstehen.
Abbildung 2. Generische Beziehung zwischen jeder toxischen Reaktion und der Dosis eines umweltbedingten, chemischen oder physikalischen Mittels
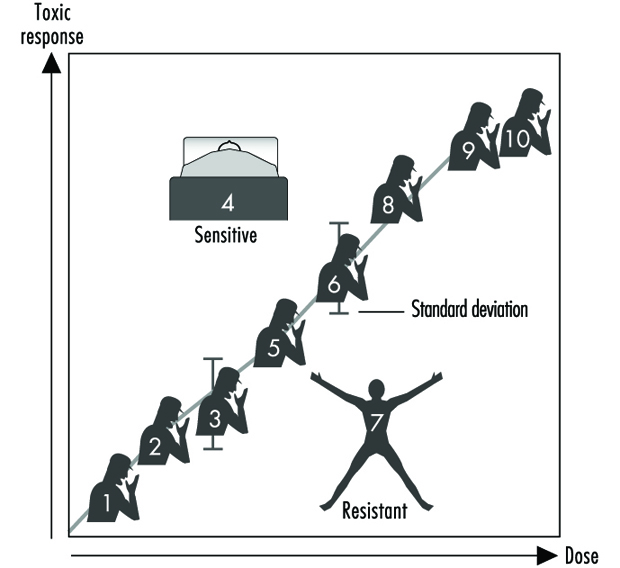
Unter Verwendung dieser Ausreißer in Familienstudien haben Wissenschaftler in einer Reihe von Laboratorien begonnen, die Bedeutung der Mendelschen Vererbung für eine bestimmte toxische Reaktion zu erkennen. Anschließend kann man sich molekularbiologischen und genetischen Studien zuwenden, um den zugrunde liegenden Mechanismus auf Genebene zu lokalisieren (Genotyp) verantwortlich für die umweltbedingte Krankheit (Phänotyp).
Xenobiotika- oder Arzneimittel-metabolisierende Enzyme
Wie reagiert der Körper auf die unzähligen exogenen Chemikalien, denen wir ausgesetzt sind? Menschen und andere Säugetiere haben hochkomplexe metabolische Enzymsysteme entwickelt, die mehr als ein Dutzend verschiedene Superfamilien von Enzymen umfassen. Nahezu jede Chemikalie, der Menschen ausgesetzt sind, wird durch diese Enzyme modifiziert, um die Entfernung der Fremdsubstanz aus dem Körper zu erleichtern. Gemeinsam werden diese Enzyme häufig als bezeichnet Arzneimittel metabolisierende Enzyme or Xenobiotika metabolisierende Enzyme. Eigentlich sind beide Begriffe irreführend. Erstens verstoffwechseln viele dieser Enzyme nicht nur Medikamente, sondern Hunderttausende von Umwelt- und Nahrungschemikalien. Zweitens haben alle diese Enzyme auch normale Körperverbindungen als Substrate; Keines dieser Enzyme verstoffwechselt nur fremde Chemikalien.
Seit mehr als vier Jahrzehnten werden die durch diese Enzyme vermittelten Stoffwechselprozesse üblicherweise entweder als Phase-I- oder als Phase-II-Reaktionen klassifiziert (Abbildung 3). Reaktionen der Phase I („Funktionalisierung“) umfassen im Allgemeinen relativ geringfügige strukturelle Modifikationen der Ausgangschemikalie durch Oxidation, Reduktion oder Hydrolyse, um einen besser wasserlöslichen Metaboliten herzustellen. Häufig bieten Phase-I-Reaktionen einen „Handgriff“ für die weitere Modifikation einer Verbindung durch nachfolgende Phase-II-Reaktionen. Phase-I-Reaktionen werden in erster Linie durch eine Superfamilie äußerst vielseitiger Enzyme vermittelt, die zusammen als Cytochrome P450 bezeichnet werden, obwohl auch andere Enzym-Superfamilien beteiligt sein können (Abbildung 4).
Abbildung 3. Die klassische Bezeichnung von Phase-I- und Phase-II-Fremdstoff- oder Arzneimittel-metabolisierenden Enzymen
Abbildung 4. Beispiele für Arzneimittel metabolisierende Enzyme
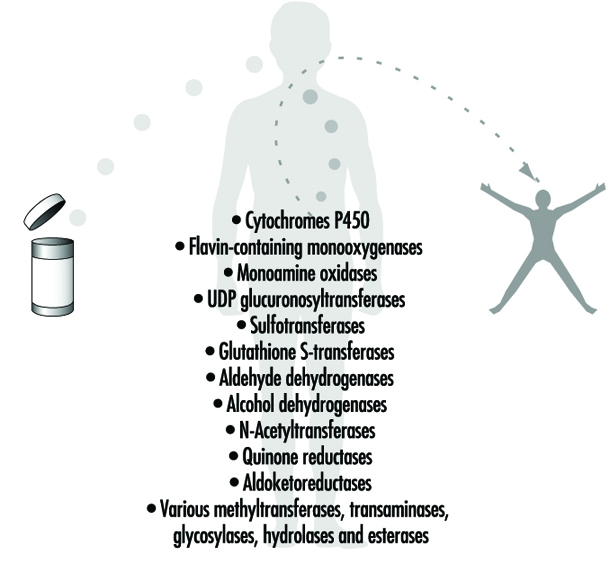
Phase-II-Reaktionen umfassen die Kopplung eines wasserlöslichen endogenen Moleküls an eine Chemikalie (Ausgangschemikalie oder Phase-I-Metabolit), um die Ausscheidung zu erleichtern. Phase-II-Reaktionen werden häufig als "Konjugations"- oder "Derivatisierungs"-Reaktionen bezeichnet. Die Enzym-Superfamilien, die Phase-II-Reaktionen katalysieren, werden im Allgemeinen nach der beteiligten endogenen konjugierenden Einheit benannt: zum Beispiel Acetylierung durch die N-Acetyltransferasen, Sulfatierung durch die Sulfotransferasen, Glutathion-Konjugation durch die Glutathion-Transferasen und Glucuronidierung durch die UDP-Glucuronosyltransferasen (Abbildung 4). . Obwohl das Hauptorgan des Arzneimittelstoffwechsels die Leber ist, sind die Konzentrationen einiger Arzneimittel metabolisierender Enzyme im Gastrointestinaltrakt, den Keimdrüsen, der Lunge, dem Gehirn und den Nieren ziemlich hoch, und solche Enzyme sind zweifellos bis zu einem gewissen Grad in jeder lebenden Zelle vorhanden.
Xenobiotika-metabolisierende Enzyme sind zweischneidig Swords
Je mehr wir über die biologischen und chemischen Prozesse erfahren, die zu Fehlentwicklungen der menschlichen Gesundheit führen, desto offensichtlicher wird, dass Arzneimittel metabolisierende Enzyme auf ambivalente Weise funktionieren (Abbildung 3). In den meisten Fällen werden fettlösliche Chemikalien in leichter ausgeschiedene wasserlösliche Metaboliten umgewandelt. Es ist jedoch klar, dass dieselben Enzyme in vielen Fällen in der Lage sind, andere inerte Chemikalien in hochreaktive Moleküle umzuwandeln. Diese Zwischenprodukte können dann mit zellulären Makromolekülen wie Proteinen und DNA interagieren. Somit besteht für jede Chemikalie, der Menschen ausgesetzt sind, das Potenzial für konkurrierende Stoffwechselwege metabolische Aktivierung und Entgiftung.
Kurze Überprüfung der Genetik
In der Humangenetik ist jedes Gen (loci) befindet sich auf einem der 23 Chromosomenpaare. Die Zwei Allele (einer auf jedem Chromosom des Paares vorhanden) können gleich oder voneinander verschieden sein. Zum Beispiel die B und b Allele, in denen B (braune Augen) dominiert b (blaue Augen): Individuen des braunäugigen Phänotyps können entweder die haben BB or Bb Genotypen, wohingegen Individuen des blauäugigen Phänotyps nur die haben können bb Genotyp.
A Polymorphismus ist definiert als zwei oder mehr stabil vererbte Phänotypen (Merkmale) – die von denselben Genen abstammen – die in der Population aufrechterhalten werden, oft aus Gründen, die nicht unbedingt offensichtlich sind. Damit ein Gen polymorph ist, darf das Genprodukt für die Entwicklung, die Fortpflanzungskraft oder andere kritische Lebensprozesse nicht wesentlich sein. Tatsächlich ist ein „ausgewogener Polymorphismus“, bei dem die Heterozygote einen deutlichen Überlebensvorteil gegenüber beiden Homozygoten hat (z. B. Resistenz gegen Malaria und das Sichelzellen-Hämoglobin-Allel), eine häufige Erklärung dafür, dass ein Allel in der Population auf einem ansonsten unerklärlichen hohen Wert gehalten wird Frequenzen (vgl González und Nebert 1990).
Menschliche Polymorphismen von Xenobiotika-metabolisierenden Enzymen
Genetische Unterschiede im Metabolismus verschiedener Medikamente und Umweltchemikalien sind seit mehr als vier Jahrzehnten bekannt (Kalow 1962 und 1992). Diese Unterschiede werden häufig als bezeichnet pharmakogenetisch oder allgemeiner ökogenetische Polymorphismen. Diese Polymorphismen stellen abweichende Allele dar, die mit einer relativ hohen Häufigkeit in der Bevölkerung vorkommen und im Allgemeinen mit Aberrationen in der Enzymexpression oder -funktion assoziiert sind. In der Vergangenheit wurden Polymorphismen normalerweise nach unerwarteten Reaktionen auf Therapeutika identifiziert. In jüngerer Zeit hat die rekombinante DNA-Technologie es Wissenschaftlern ermöglicht, die genauen Veränderungen in Genen zu identifizieren, die für einige dieser Polymorphismen verantwortlich sind. Polymorphismen wurden nun in vielen Arzneimittel metabolisierenden Enzymen charakterisiert – einschließlich sowohl Phase-I- als auch Phase-II-Enzymen. Da immer mehr Polymorphismen identifiziert werden, wird es immer deutlicher, dass jedes Individuum ein bestimmtes Komplement von Arzneimittel-metabolisierenden Enzymen besitzen kann. Diese Vielfalt könnte man als „metabolischen Fingerabdruck“ bezeichnen. Es ist das komplexe Zusammenspiel der verschiedenen Superfamilien der arzneimittelmetabolisierenden Enzyme in jedem Individuum, das letztendlich seine oder ihre besondere Reaktion auf eine bestimmte Chemikalie bestimmt (Kalow 1962 und 1992; Nebert 1988; Gonzalez und Nebert 1990; Nebert und Weber 1990).
Expression menschlicher Xenobiotika-metabolisierender Enzyme in der Zelle KULTUR
Wie könnten wir bessere Prädiktoren für menschliche toxische Reaktionen auf Chemikalien entwickeln? Fortschritte bei der Definition der Vielzahl von Enzymen, die Arzneimittel metabolisieren, müssen von genauen Kenntnissen darüber begleitet werden, welche Enzyme das Stoffwechselschicksal einzelner Chemikalien bestimmen. Daten aus Laborstudien an Nagetieren haben sicherlich nützliche Informationen geliefert. Signifikante Unterschiede zwischen den Spezies bei Enzymen, die Fremdstoffe metabolisieren, erfordern jedoch Vorsicht bei der Extrapolation von Daten auf menschliche Populationen. Um diese Schwierigkeit zu überwinden, haben viele Laboratorien Systeme entwickelt, in denen verschiedene Zelllinien in Kultur manipuliert werden können, um funktionelle menschliche Enzyme zu produzieren, die stabil und in hohen Konzentrationen sind (Gonzalez, Crespi und Gelboin 1991). Die erfolgreiche Produktion menschlicher Enzyme wurde in einer Vielzahl unterschiedlicher Zelllinien aus Quellen wie Bakterien, Hefen, Insekten und Säugetieren erreicht.
Um den Stoffwechsel von Chemikalien noch genauer zu definieren, mehrere Enzyme wurden auch erfolgreich in einer einzelnen Zelllinie produziert (Gonzalez, Crespi und Gelboin 1991). Solche Zelllinien liefern wertvolle Einblicke in die genauen Enzyme, die an der metabolischen Verarbeitung einer bestimmten Verbindung und wahrscheinlich toxischer Metaboliten beteiligt sind. Wenn diese Informationen dann mit dem Wissen über das Vorhandensein und die Menge eines Enzyms in menschlichen Geweben kombiniert werden können, sollten diese Daten wertvolle Prädiktoren für das Ansprechen liefern.
Cytochrom P450
Geschichte und Nomenklatur
Die Cytochrom-P450-Superfamilie ist eine der am besten untersuchten Superfamilien von Arzneimittel metabolisierenden Enzymen, die eine große individuelle Variabilität in der Reaktion auf Chemikalien aufweist. Cytochrom P450 ist ein praktischer Oberbegriff, der verwendet wird, um eine große Superfamilie von Enzymen zu beschreiben, die für den Metabolismus unzähliger endogener und exogener Substrate von entscheidender Bedeutung sind. Der Begriff Cytochrom P450 wurde erstmals 1962 geprägt, um ein Unbekanntes zu beschreiben Pigment in Zellen, die, wenn sie reduziert und mit Kohlenmonoxid gebunden wurden, einen charakteristischen Absorptionspeak bei 450 nm erzeugten. Seit den frühen 1980er Jahren hat die cDNA-Klonierungstechnologie zu bemerkenswerten Einblicken in die Vielzahl von Cytochrom-P450-Enzymen geführt. Bis heute wurden mehr als 400 unterschiedliche Cytochrom-P450-Gene in Tieren, Pflanzen, Bakterien und Hefen identifiziert. Es wurde geschätzt, dass jede Säugetierart, wie der Mensch, 60 oder mehr unterschiedliche P450-Gene besitzen kann (Nebert und Nelson 1991). Die Vielzahl der P450-Gene hat die Entwicklung eines standardisierten Nomenklatursystems erforderlich gemacht (Nebert et al. 1987; Nelson et al. 1993). Das erstmals 1987 vorgeschlagene und alle zwei Jahre aktualisierte Nomenklatursystem basiert auf der divergenten Entwicklung von Aminosäuresequenzvergleichen zwischen P450-Proteinen. Die P450-Gene werden in Familien und Unterfamilien unterteilt: Enzyme innerhalb einer Familie zeigen mehr als 40 % Aminosäureähnlichkeit, und diejenigen innerhalb derselben Unterfamilie zeigen 55 % Ähnlichkeit. P450-Gene werden mit dem Wurzelsymbol benannt CYP gefolgt von einer arabischen Zahl, die die P450-Familie bezeichnet, einem Buchstaben, der die Unterfamilie bezeichnet, und einer weiteren arabischen Zahl, die das individuelle Gen bezeichnet (Nelson et al. 1993; Nebert et al. 1991). Daher, CYP1A1 repräsentiert das P450-Gen 1 in Familie 1 und Unterfamilie A.
Ab Februar 1995 gibt es 403 CYP Gene in der Datenbank, bestehend aus 59 Familien und 105 Unterfamilien. Dazu gehören acht niedere eukaryotische Familien, 15 Pflanzenfamilien und 19 Bakterienfamilien. Die 15 menschlichen P450-Genfamilien umfassen 26 Unterfamilien, von denen 22 auf chromosomale Stellen im größten Teil des Genoms kartiert wurden. Einige Sequenzen sind über viele Arten eindeutig ortholog – zum Beispiel nur eine CYP17 (Steroid 17α-Hydroxylase)-Gen wurde in allen bisher untersuchten Wirbeltieren gefunden; andere Sequenzen innerhalb einer Unterfamilie sind stark dupliziert, was die Identifizierung orthologer Paare unmöglich macht (z. B. die CYP2C Unterfamilie). Interessanterweise teilen Mensch und Hefe ein orthologes Gen in der CYP51 Familie. Für Leser, die weitere Informationen über die P450-Superfamilie suchen, stehen zahlreiche umfassende Übersichtsartikel zur Verfügung (Nelson et al. 1993; Nebert et al. 1991; Nebert und McKinnon 1994; Guengerich 1993; Gonzalez 1992).
Der Erfolg des P450-Nomenklatursystems hat zur Entwicklung ähnlicher Terminologiesysteme für die UDP-Glucuronosyltransferasen (Burchell et al. 1991) und Flavin-enthaltende Monooxygenasen (Lawton et al. 1994) geführt. Ähnliche Nomenklatursysteme, die auf divergenter Evolution basieren, werden auch für mehrere andere Superfamilien von Arzneimittel metabolisierenden Enzymen entwickelt (z. B. Sulfotransferasen, Epoxidhydrolasen und Aldehyddehydrogenasen).
Kürzlich teilten wir die Säuger-P450-Gen-Superfamilie in drei Gruppen ein (Nebert und McKinnon 1994) – diejenigen, die hauptsächlich am Stoffwechsel von Fremdchemikalien beteiligt sind, diejenigen, die an der Synthese verschiedener Steroidhormone beteiligt sind, und diejenigen, die an anderen wichtigen endogenen Funktionen beteiligt sind. Es sind die fremdstoffmetabolisierenden P450-Enzyme, denen die größte Bedeutung für die Vorhersage der Toxizität zukommt.
Xenobiotika metabolisierende P450-Enzyme
P450-Enzyme, die am Metabolismus fremder Verbindungen und Arzneimittel beteiligt sind, kommen fast immer innerhalb von Familien vor CYP1, CYP2, CYP3 und CYP4. Diese P450-Enzyme katalysieren eine Vielzahl von Stoffwechselreaktionen, wobei ein einziges P450 oft in der Lage ist, viele verschiedene Verbindungen zu metabolisieren. Darüber hinaus können mehrere P450-Enzyme eine einzelne Verbindung an verschiedenen Stellen metabolisieren. Eine Verbindung kann auch an derselben einzelnen Stelle von mehreren P450 metabolisiert werden, wenn auch mit unterschiedlichen Raten.
Eine äußerst wichtige Eigenschaft der Arzneimittel metabolisierenden P450-Enzyme besteht darin, dass viele dieser Gene durch genau die Substanzen induzierbar sind, die als ihre Substrate dienen. Andererseits werden andere P450-Gene durch Nichtsubstrate induziert. Dieses Phänomen der Enzyminduktion liegt vielen Arzneimittelwechselwirkungen von therapeutischer Bedeutung zugrunde.
Obwohl diese speziellen P450-Enzyme in vielen Geweben vorhanden sind, werden sie in relativ hohen Konzentrationen in der Leber, dem primären Ort des Arzneimittelstoffwechsels, gefunden. Einige der Xenobiotika metabolisierenden P450-Enzyme zeigen Aktivität gegenüber bestimmten endogenen Substraten (z. B. Arachidonsäure). Es wird jedoch allgemein angenommen, dass die meisten dieser Xenobiotika metabolisierenden P450-Enzyme keine wichtige physiologische Rolle spielen – obwohl dies noch nicht experimentell nachgewiesen wurde. Die selektive homozygote Disruption oder „Knock-out“ einzelner Xenobiotika-metabolisierender P450-Gene mittels Gen-Targeting-Methoden in Mäusen wird wahrscheinlich bald eindeutige Informationen in Bezug auf die physiologische Rolle der Xenobiotika-metabolisierenden P450s liefern (für eine Übersicht von Gen-Targeting, siehe Capecchi 1994).
Im Gegensatz zu P450-Familien, die für Enzyme codieren, die hauptsächlich an physiologischen Prozessen beteiligt sind, zeigen Familien, die Fremdstoffe metabolisierende P450-Enzyme codieren, eine ausgeprägte Artspezifität und enthalten häufig viele aktive Gene pro Unterfamilie (Nelson et al. 1993; Nebert et al. 1991). Angesichts des offensichtlichen Mangels an physiologischen Substraten ist es möglich, dass P450-Enzyme in Familien vorkommen CYP1, CYP2, CYP3 und CYP4 die in den letzten mehreren hundert Millionen Jahren erschienen sind, haben sich als Mittel zur Entgiftung von Fremdchemikalien entwickelt, die in der Umwelt und in der Ernährung angetroffen werden. Die Evolution der Xenobiotika metabolisierenden P450 hätte eindeutig über einen Zeitraum stattgefunden, der weit vor der Synthese der meisten synthetischen Chemikalien liegt, denen Menschen heute ausgesetzt sind. Die Gene in diesen vier Genfamilien könnten sich in Tieren aufgrund ihrer Exposition gegenüber pflanzlichen Metaboliten während der letzten 1.2 Milliarden Jahre entwickelt und auseinander entwickelt haben – ein Prozess, der beschreibend als „Tier-Pflanze-Kriegsführung“ bezeichnet wird (Gonzalez und Nebert 1990). Tier-Pflanze-Krieg ist das Phänomen, bei dem Pflanzen neue Chemikalien (Phytoalexine) als Abwehrmechanismus entwickelten, um die Aufnahme durch Tiere zu verhindern, und die Tiere wiederum mit der Entwicklung neuer P450-Gene reagierten, um sich an die sich diversifizierenden Substrate anzupassen. Weitere Impulse für diesen Vorschlag liefern die kürzlich beschriebenen Beispiele von Pflanzen-Insekten- und Pflanzen-Pilz-Chemical Warfare, die P450-Entgiftung von toxischen Substraten beinhalten (Nebert 1994).
Das Folgende ist eine kurze Einführung in mehrere der Human-Xenobiotika-metabolisierenden P450-Enzym-Polymorphismen, bei denen angenommen wird, dass genetische Determinanten der toxischen Reaktion von hoher Bedeutung sind. Bis vor kurzem wurden P450-Polymorphismen im Allgemeinen durch unerwartete Schwankungen in der Patientenreaktion auf verabreichte therapeutische Mittel nahegelegt. Mehrere P450-Polymorphismen werden tatsächlich nach dem Medikament benannt, mit dem der Polymorphismus zuerst identifiziert wurde. In jüngerer Zeit konzentrierten sich Forschungsbemühungen auf die Identifizierung der genauen P450-Enzyme, die am Stoffwechsel von Chemikalien beteiligt sind, für die eine Varianz beobachtet wird, und auf die genaue Charakterisierung der beteiligten P450-Gene. Wie zuvor beschrieben, kann die messbare Aktivität eines P450-Enzyms gegenüber einer Modellchemikalie als Phänotyp bezeichnet werden. Allele Unterschiede in einem P450-Gen für jedes Individuum werden als P450-Genotyp bezeichnet. Da die Analyse von P450-Genen immer genauer untersucht wird, wird die genaue molekulare Grundlage der zuvor dokumentierten phänotypischen Varianz immer klarer.
Die CYP1A-Unterfamilie
Das CYP1A Unterfamilie umfasst zwei Enzyme in Menschen und allen anderen Säugetieren: diese werden unter der Standard-P1-Nomenklatur als CYP1A1 und CYP2A450 bezeichnet. Diese Enzyme sind von erheblichem Interesse, da sie an der metabolischen Aktivierung vieler Prokarzinogene beteiligt sind und auch durch mehrere toxikologisch bedenkliche Verbindungen, einschließlich Dioxin, induziert werden. Zum Beispiel aktiviert CYP1A1 metabolisch viele Verbindungen, die im Zigarettenrauch gefunden werden. CYP1A2 aktiviert metabolisch viele Arylamine, die mit Harnblasenkrebs in Verbindung gebracht werden und in der chemischen Farbstoffindustrie vorkommen. CYP1A2 aktiviert auch metabolisch 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanon (NNK), ein aus Tabak gewonnenes Nitrosamin. CYP1A1 und CYP1A2 werden aufgrund der Induktion durch im Rauch vorhandene polyzyklische Kohlenwasserstoffe auch in höheren Konzentrationen in den Lungen von Zigarettenrauchern gefunden. Die Niveaus der CYP1A1- und CYP1A2-Aktivität gelten daher als wichtige Determinanten der individuellen Reaktion auf viele potenziell toxische Chemikalien.
Toxikologisches Interesse an der CYP1A Unterfamilie wurde durch einen Bericht aus dem Jahr 1973 stark intensiviert, der den Grad der CYP1A1-Induzierbarkeit bei Zigarettenrauchern mit der individuellen Anfälligkeit für Lungenkrebs korrelierte (Kellermann, Shaw und Luyten-Kellermann 1973). Die molekularen Grundlagen der CYP1A1- und CYP1A2-Induktion standen im Mittelpunkt zahlreicher Labors. Der Induktionsprozess wird durch ein als Ah-Rezeptor bezeichnetes Protein vermittelt, an das Dioxine und strukturell verwandte Chemikalien binden. Der Name Ah ist abgeleitet von der aryl hKohlenwasserstoffnatur vieler CYP1A-Induktoren. Interessanterweise führen Unterschiede in dem Gen, das den Ah-Rezeptor codiert, zwischen Mäusestämmen zu deutlichen Unterschieden in der chemischen Reaktion und Toxizität. Ein Polymorphismus im Ah-Rezeptorgen scheint auch beim Menschen vorzukommen: Etwa ein Zehntel der Bevölkerung zeigt eine hohe Induktion von CYP1A1 und ist möglicherweise einem größeren Risiko als die anderen neun Zehntel der Bevölkerung für die Entwicklung bestimmter chemisch induzierter Krebsarten ausgesetzt. Die Rolle des Ah-Rezeptors bei der Kontrolle von Enzymen in der CYP1A Subfamilie und ihre Rolle als Determinante der menschlichen Reaktion auf Chemikalienexposition waren Gegenstand mehrerer neuerer Übersichten (Nebert, Petersen und Puga 1991; Nebert, Puga und Vasiliou 1993).
Gibt es andere Polymorphismen, die den Gehalt an CYP1A-Proteinen in einer Zelle kontrollieren könnten? Ein Polymorphismus in der CYP1A1 Gen wurde ebenfalls identifiziert, und dies scheint das Lungenkrebsrisiko bei japanischen Zigarettenrauchern zu beeinflussen, obwohl derselbe Polymorphismus das Risiko bei anderen ethnischen Gruppen nicht zu beeinflussen scheint (Nebert und McKinnon 1994).
CYP2C19
Schwankungen in der Rate, mit der Individuen das Antikonvulsivum (S)-Mephenytoin metabolisieren, sind seit vielen Jahren gut dokumentiert (Guengerich 1989). Zwischen 2 % und 5 % der Kaukasier und bis zu 25 % der Asiaten weisen einen Mangel an dieser Aktivität auf und sind möglicherweise einem höheren Toxizitätsrisiko durch das Medikament ausgesetzt. Es ist seit langem bekannt, dass dieser Enzymdefekt ein Mitglied des Menschen betrifft CYP2C Unterfamilie, aber die genaue molekulare Grundlage dieses Mangels war Gegenstand beträchtlicher Kontroversen. Der Hauptgrund für diese Schwierigkeit waren die sechs oder mehr Gene im Menschen CYP2C Unterfamilie. Kürzlich wurde jedoch gezeigt, dass eine Einzelbasen-Mutation in der CYP2C19 -Gen ist die Hauptursache für diesen Mangel (Goldstein und de Morais 1994). Ein einfacher DNA-Test, basierend auf der Polymerase-Kettenreaktion (PCR), wurde ebenfalls entwickelt, um diese Mutation schnell in menschlichen Populationen zu identifizieren (Goldstein und de Morais 1994).
CYP2D6
Die vielleicht am ausführlichsten charakterisierte Variation in einem P450-Gen ist diejenige, an der die beteiligt ist CYP2D6 Gen. Mehr als ein Dutzend Beispiele für Mutationen, Umlagerungen und Deletionen, die dieses Gen betreffen, wurden beschrieben (Meyer 1994). Dieser Polymorphismus wurde erstmals vor 20 Jahren durch die klinische Variabilität der Reaktion der Patienten auf das Antihypertensivum Debrisoquin nahegelegt. Änderungen in der CYP2D6 Gene, die zu einer veränderten Enzymaktivität führen, werden daher zusammenfassend als die bezeichnet Debrisoquiner Polymorphismus.
Vor dem Aufkommen von DNA-basierten Studien wurden Personen basierend auf Metabolitenkonzentrationen in Urinproben als langsame oder extensive Metabolisierer (PMs, EMs) von Debrisoquin klassifiziert. Es ist jetzt klar, dass Änderungen in der CYP2D6 -Gen kann dazu führen, dass Individuen nicht nur einen schlechten oder ausgedehnten Debrisoquin-Metabolismus, sondern auch einen ultraschnellen Metabolismus aufweisen. Die meisten Änderungen in der CYP2D6 Gen sind mit einem teilweisen oder vollständigen Mangel an Enzymfunktion verbunden; Kürzlich wurden jedoch Personen in zwei Familien beschrieben, die mehrere funktionelle Kopien des besitzen CYP2D6 Gen, was zu einem ultraschnellen Metabolismus von CYP2D6-Substraten führt (Meyer 1994). Diese bemerkenswerte Beobachtung liefert neue Einblicke in das breite Spektrum der CYP2D6-Aktivität, die zuvor in Bevölkerungsstudien beobachtet wurde. Veränderungen der CYP2D6-Funktion sind angesichts der mehr als 30 häufig verschriebenen Medikamente, die von diesem Enzym metabolisiert werden, von besonderer Bedeutung. Die CYP2D6-Funktion eines Individuums ist daher eine Hauptdeterminante sowohl der therapeutischen als auch der toxischen Reaktion auf die verabreichte Therapie. In der Tat wurde kürzlich argumentiert, dass die Berücksichtigung des CYP2D6-Status eines Patienten für die sichere Anwendung von psychiatrischen und kardiovaskulären Arzneimitteln notwendig ist.
Die Rolle des CYP2D6 Polymorphismus als Determinante der individuellen Anfälligkeit für menschliche Krankheiten wie Lungenkrebs und Parkinson-Krankheit war ebenfalls Gegenstand intensiver Studien (Nebert und McKinnon 1994; Meyer 1994). Während Schlussfolgerungen angesichts der unterschiedlichen Art der verwendeten Studienprotokolle schwer zu definieren sind, scheinen die meisten Studien auf einen Zusammenhang zwischen schnellen Metabolisierern von Debrisoquin (EM-Phänotyp) und Lungenkrebs hinzuweisen. Die Gründe für eine solche Assoziation sind derzeit unklar. Es wurde jedoch gezeigt, dass das CYP2D6-Enzym NNK, ein aus Tabak gewonnenes Nitrosamin, metabolisiert.
Mit der Verbesserung DNA-basierter Assays, die eine noch genauere Beurteilung des CYP2D6-Status ermöglichen, wird erwartet, dass die genaue Beziehung von CYP2D6 zum Krankheitsrisiko geklärt wird. Während der schnelle Metabolisierer mit einer Anfälligkeit für Lungenkrebs in Verbindung gebracht werden kann, scheint der langsame Metabolisierer (PM-Phänotyp) mit der Parkinson-Krankheit unbekannter Ursache assoziiert zu sein. Während diese Studien auch schwer zu vergleichen sind, scheint es, dass PM-Personen mit einer verminderten Fähigkeit, CYP2D6-Substrate (z. B. Debrisoquin) zu metabolisieren, ein 2- bis 2.5-fach erhöhtes Risiko haben, an der Parkinson-Krankheit zu erkranken.
CYP2E1
Das CYP2E1 Das Gen kodiert für ein Enzym, das viele Chemikalien verstoffwechselt, darunter Medikamente und viele niedermolekulare Karzinogene. Dieses Enzym ist auch deshalb von Interesse, weil es durch Alkohol stark induzierbar ist und eine Rolle bei Leberschäden spielen kann, die durch Chemikalien wie Chloroform, Vinylchlorid und Tetrachlorkohlenstoff verursacht werden. Das Enzym wird hauptsächlich in der Leber gefunden, und der Enzymspiegel variiert deutlich zwischen Individuen. Genaue Prüfung der CYP2E1 -Gen hat zur Identifizierung mehrerer Polymorphismen geführt (Nebert und McKinnon 1994). Es wurde eine Beziehung zwischen dem Vorhandensein bestimmter struktureller Variationen in der CYP2E1 Gen und offensichtlich verringertes Lungenkrebsrisiko in einigen Studien; Es gibt jedoch deutliche interethnische Unterschiede, die einer Klärung dieser möglichen Beziehung bedürfen.
Die CYP3A-Unterfamilie
Beim Menschen wurden vier Enzyme als Mitglieder der identifiziert CYP3A Unterfamilie aufgrund ihrer Ähnlichkeit in der Aminosäuresequenz. Die CYP3A-Enzyme metabolisieren viele häufig verschriebene Medikamente wie Erythromycin und Cyclosporin. Der krebserregende Lebensmittelkontaminant Aflatoxin B1 ist ebenfalls ein CYP3A-Substrat. Ein Mitglied des Menschen CYP3A Unterfamilie, bezeichnet CYP3A4, ist das wichtigste P450 in der menschlichen Leber und kommt auch im Gastrointestinaltrakt vor. Wie bei vielen anderen P450-Enzymen ist der CYP3A4-Spiegel von Person zu Person sehr unterschiedlich. Ein zweites Enzym mit der Bezeichnung CYP3A5 findet sich nur in etwa 25 % der Lebern; die genetische Grundlage dieses Befundes wurde nicht aufgeklärt. Die Bedeutung der CYP3A4- oder CYP3A5-Variabilität als Faktor für genetische Determinanten der toxischen Reaktion wurde noch nicht nachgewiesen (Nebert und McKinnon 1994).
Nicht-P450-Polymorphismen
Zahlreiche Polymorphismen existieren auch innerhalb anderer Xenobiotika-metabolisierender Enzym-Superfamilien (z. B. Glutathiontransferasen, UDP-Glucuronosyltransferasen, para-Oxonasen, Dehydrogenasen, N-Acetyltransferasen und Flavin-enthaltende Monooxygenasen). Da die endgültige Toxizität jedes durch P450 erzeugten Zwischenprodukts von der Effizienz nachfolgender Phase-II-Entgiftungsreaktionen abhängt, ist die kombinierte Rolle mehrerer Enzympolymorphismen wichtig bei der Bestimmung der Anfälligkeit für chemisch induzierte Krankheiten. Das metabolische Gleichgewicht zwischen Phase-I- und Phase-II-Reaktionen (Abbildung 3) ist daher wahrscheinlich ein Hauptfaktor bei chemisch induzierten menschlichen Krankheiten und genetischen Determinanten der toxischen Reaktion.
Der GSTM1-Genpolymorphismus
Ein gut untersuchtes Beispiel eines Polymorphismus in einem Phase-II-Enzym ist dasjenige, an dem ein Mitglied der Glutathion-S-Transferase-Enzymsuperfamilie mit der Bezeichnung GST mu oder GSTM1 beteiligt ist. Dieses spezielle Enzym ist von erheblichem toxikologischem Interesse, da es anscheinend an der anschließenden Entgiftung toxischer Metaboliten beteiligt ist, die aus Chemikalien im Zigarettenrauch durch das CYP1A1-Enzym produziert werden. Der identifizierte Polymorphismus in diesem Glutathion-Transferase-Gen beinhaltet ein völliges Fehlen eines funktionellen Enzyms bei nicht weniger als der Hälfte aller untersuchten Kaukasier. Dieses Fehlen eines Phase-II-Enzyms scheint mit einer erhöhten Anfälligkeit für Lungenkrebs verbunden zu sein. Durch die Gruppierung von Personen nach beiden Varianten CYP1A1 Gene und die Deletion oder Anwesenheit eines funktionellen GSM1 Gens wurde gezeigt, dass das Risiko, an rauchinduziertem Lungenkrebs zu erkranken, signifikant variiert (Kawajiri, Watanabe und Hayashi 1994). Insbesondere Personen, die eine Seltenheit zeigen CYP1A1 Genveränderung, in Kombination mit einem Fehlen der GSM1 Gen, hatten ein höheres Risiko (bis zu neunfach) an Lungenkrebs zu erkranken, wenn sie einer relativ geringen Menge an Zigarettenrauch ausgesetzt waren. Interessanterweise scheint es interethnische Unterschiede in der Bedeutung von Genvarianten zu geben, die weitere Untersuchungen erfordern, um die genaue Rolle solcher Veränderungen bei der Krankheitsanfälligkeit aufzuklären (Kalow 1962; Nebert und McKinnon 1994; Kawajiri, Watanabe und Hayashi 1994).
Synergistischer Effekt von zwei oder mehr Polymorphismen auf die Toxizität Antwort
Eine toxische Reaktion auf ein Umweltmittel kann durch die Kombination zweier pharmakogenetischer Defekte bei demselben Individuum stark übertrieben werden, beispielsweise die kombinierten Wirkungen des N-Acetyltransferase (NAT2)-Polymorphismus und des Glucose-6-Phosphat-Dehydrogenase (G6PD)-Polymorphismus .
Die berufliche Exposition gegenüber Arylaminen stellt ein ernstes Risiko für Harnblasenkrebs dar. Seit den eleganten Studien von Cartwright im Jahr 1954 ist klar geworden, dass der N-Acetylator-Status eine Determinante von Azofarbstoff-induziertem Blasenkrebs ist. Es besteht eine hochsignifikante Korrelation zwischen dem Slow-Acetylator-Phänotyp und dem Auftreten von Blasenkrebs sowie dem Grad der Invasivität dieses Krebses in der Blasenwand. Im Gegenteil, es besteht eine signifikante Assoziation zwischen dem Phänotyp des schnellen Acetylierers und dem Auftreten von kolorektalen Karzinomen. Die N-Acetyltransferase (NAT1, NAT2)-Gene wurden geklont und sequenziert, und DNA-basierte Assays sind nun in der Lage, mehr als ein Dutzend allelische Varianten nachzuweisen, die für den Phänotyp des langsamen Acetylierers verantwortlich sind. Das NAT2 Das Gen ist polymorph und für den größten Teil der Variabilität der toxischen Reaktion auf Umweltchemikalien verantwortlich (Weber 1987; Grant 1993).
Glucose-6-Phosphat-Dehydrogenase (G6PD) ist ein Enzym, das für die Bildung und Aufrechterhaltung von NADPH entscheidend ist. Niedrige oder fehlende G6PD-Aktivität kann aufgrund des Fehlens normaler Spiegel von reduziertem Glutathion (GSH) in den roten Blutkörperchen zu schwerer arzneimittel- oder xenobiotikainduzierter Hämolyse führen. G6PD-Mangel betrifft weltweit mindestens 300 Millionen Menschen. Mehr als 10 % der afroamerikanischen Männer weisen den weniger schweren Phänotyp auf, während bestimmte sardische Gemeinschaften den schwereren „mediterranen Typ“ mit einer Häufigkeit von bis zu einer von drei Personen aufweisen. Das G6PD Das Gen wurde geklont und auf dem X-Chromosom lokalisiert, und zahlreiche verschiedene Punktmutationen sind für den großen Grad an phänotypischer Heterogenität verantwortlich, die bei G6PD-defizienten Individuen beobachtet wird (Beutler 1992).
Es wurde festgestellt, dass Thiozalsulfon, ein Arylamin-Sulfat-Medikament, eine bimodale Verteilung der hämolytischen Anämie in der behandelten Population verursacht. Bei der Behandlung mit bestimmten Arzneimitteln sind Personen mit der Kombination aus G6PD-Mangel und dem langsamen Acetylierer-Phänotyp stärker betroffen als Personen mit dem G6PD-Mangel allein oder dem langsamen Acetylierer-Phänotyp allein. G6PD-defiziente langsame Acetylierer sind mindestens 40-mal anfälliger für Thiozalsulfon-induzierte Hämolyse als normale G6PD-schnelle Acetylierer.
Wirkung genetischer Polymorphismen auf die Expositionsabschätzung
Die Expositionsabschätzung und das Biomonitoring (Abbildung 1) erfordern auch Informationen über die genetische Ausstattung jedes Individuums. Bei identischer Exposition gegenüber einer gefährlichen Chemikalie kann der Gehalt an Hämoglobin-Addukten (oder anderen Biomarkern) zwischen Personen um zwei oder drei Größenordnungen variieren, abhängig vom metabolischen Fingerabdruck jeder Person.
Dieselbe kombinierte Pharmakogenetik wurde bei Arbeitern in Chemiefabriken in Deutschland untersucht (Tabelle 1). Hämoglobin-Addukte bei Arbeitern, die Anilin und Acetanilid ausgesetzt waren, sind bei langsamen Acetylierern mit G6PD-Mangel im Vergleich zu den anderen möglichen kombinierten pharmakogenetischen Phänotypen bei weitem am höchsten. Diese Studie hat wichtige Implikationen für die Expositionsbewertung. Diese Daten zeigen, dass, obwohl zwei Personen am Arbeitsplatz möglicherweise der gleichen Umgebungskonzentration gefährlicher Chemikalien ausgesetzt sind, die Höhe der Exposition (über Biomarker wie Hämoglobinaddukte) auf zwei oder mehr Größenordnungen geringer geschätzt werden kann auf die zugrunde liegende genetische Veranlagung des Individuums. Ebenso kann das resultierende Risiko einer gesundheitlichen Beeinträchtigung um zwei oder mehr Größenordnungen variieren.
Tabelle 1: Hämoglobinaddukte bei Arbeitern, die gegenüber Anilin und Acetanilid exponiert waren
| Acetylator-Status | G6PD-Mangel | |||
| Schnell | Bremst | Nein | Ja | Hgb-Addukte |
| + | + | 2 | ||
| + | + | 30 | ||
| + | + | 20 | ||
| + | + | 100 | ||
Quelle: Adaptiert von Lewalter und Korallus 1985.
Genetische Unterschiede in der Bindung sowie im Stoffwechsel
Es sollte betont werden, dass die gleichen Argumente, die hier für den Metabolismus gemacht wurden, auch für die Bindung gemacht werden können. Vererbbare Unterschiede in der Bindung von Umweltmitteln werden die toxische Reaktion stark beeinflussen. Zum Beispiel Unterschiede in der Maus cdm -Gen kann die individuelle Empfindlichkeit gegenüber Cadmium-induzierter Hodennekrose tiefgreifend beeinflussen (Taylor, Heiniger und Meier 1973). Unterschiede in der Bindungsaffinität des Ah-Rezeptors wirken sich wahrscheinlich auf dioxininduzierte Toxizität und Krebs aus (Nebert, Petersen und Puga 1991; Nebert, Puga und Vasiliou 1993).
Abbildung 5 fasst die Rolle des Metabolismus und der Bindung bei Toxizität und Krebs zusammen. Toxische Stoffe, wie sie in der Umwelt oder nach Metabolisierung oder Bindung vorkommen, lösen ihre Wirkungen entweder über einen genotoxischen Weg (bei dem Schäden an der DNA auftreten) oder einen nicht-genotoxischen Weg (bei dem DNA-Schäden und Mutagenese nicht auftreten müssen) aus. Interessanterweise wurde kürzlich klar, dass „klassische“ DNA-schädigende Mittel über einen von reduziertem Glutathion (GSH) abhängigen nichtgenotoxischen Signaltransduktionsweg wirken können, der in Abwesenheit von DNA und außerhalb des Zellkerns auf oder nahe der Zelloberfläche initiiert wird (Devary et al. 1993). Genetische Unterschiede im Metabolismus und in der Bindung bleiben jedoch die Hauptdeterminanten bei der Kontrolle unterschiedlicher individueller toxischer Reaktionen.
Abbildung 5. Die allgemeinen Mittel, durch die Toxizität auftritt

Rolle von Arzneimittel-metabolisierenden Enzymen in der Zellfunktion
Genetisch basierte Variationen in der Funktion von Arzneimittel metabolisierenden Enzymen sind von großer Bedeutung bei der Bestimmung der individuellen Reaktion auf Chemikalien. Diese Enzyme sind entscheidend für die Bestimmung des Schicksals und des Zeitverlaufs einer fremden Chemikalie nach der Exposition.
Wie in Abbildung 5 dargestellt, kann die Bedeutung von Arzneimittel metabolisierenden Enzymen für die individuelle Anfälligkeit gegenüber Chemikalienexposition tatsächlich ein weitaus komplexeres Problem darstellen, als aus dieser einfachen Diskussion des Xenobiotika-Stoffwechsels hervorgeht. Mit anderen Worten, während der letzten zwei Jahrzehnte wurden genotoxische Mechanismen (Messungen von DNA-Addukten und Proteinaddukten) stark betont. Was aber, wenn nicht-genotoxische Mechanismen bei der Auslösung toxischer Reaktionen mindestens genauso wichtig sind wie genotoxische Mechanismen?
Wie bereits erwähnt, sind die physiologischen Rollen vieler Arzneimittel metabolisierender Enzyme, die am Xenobiotika-Metabolismus beteiligt sind, nicht genau definiert worden. Nebert (1994) hat vorgeschlagen, dass aufgrund ihrer Anwesenheit auf diesem Planeten für mehr als 3.5 Milliarden Jahre Arzneimittel-metabolisierende Enzyme ursprünglich (und sind es heute immer noch hauptsächlich) für die Regulierung der zellulären Spiegel vieler Nicht-Peptid-Liganden verantwortlich waren, die für die Transkriptionsaktivierung wichtig sind von Genen, die Wachstum, Differenzierung, Apoptose, Homöostase und neuroendokrine Funktionen beeinflussen. Darüber hinaus tritt die Toxizität der meisten, wenn nicht aller Umweltstoffe auf Agonisten or Antagonist Wirkung auf diese Signaltransduktionswege (Nebert 1994). Auf der Grundlage dieser Hypothese kann die genetische Variabilität in Arzneimittel metabolisierenden Enzymen ziemlich dramatische Auswirkungen auf viele kritische biochemische Prozesse innerhalb der Zelle haben, was zu wichtigen Unterschieden in der toxischen Reaktion führt. Es ist in der Tat möglich, dass ein solches Szenario auch vielen idiosynkratischen Nebenwirkungen zugrunde liegt, die bei Patienten auftreten, die häufig verschriebene Medikamente einnehmen.
Schlussfolgerungen
Das letzte Jahrzehnt hat bemerkenswerte Fortschritte in unserem Verständnis der genetischen Grundlage unterschiedlicher Reaktionen auf Chemikalien in Arzneimitteln, Nahrungsmitteln und Umweltschadstoffen gebracht. Arzneimittel metabolisierende Enzyme haben einen tiefgreifenden Einfluss darauf, wie Menschen auf Chemikalien reagieren. Da sich unser Bewusstsein für die Vielzahl von Enzymen, die Arzneimittel metabolisieren, weiter entwickelt, sind wir zunehmend in der Lage, das toxische Risiko für viele Arzneimittel und Umweltchemikalien besser einzuschätzen. Dies wird vielleicht am deutlichsten im Fall des CYP2D6-Cytochrom-P450-Enzyms veranschaulicht. Unter Verwendung relativ einfacher DNA-basierter Tests ist es möglich, die wahrscheinliche Reaktion eines Medikaments vorherzusagen, das hauptsächlich durch dieses Enzym metabolisiert wird; Diese Vorhersage wird die sicherere Verwendung wertvoller, aber potenziell toxischer Medikamente gewährleisten.
Die Identifizierung weiterer Polymorphismen (Phänotypen) von arzneimittelmetabolisierenden Enzymen wird in Zukunft zweifelsohne explodieren. Diese Informationen werden von verbesserten, minimalinvasiven DNA-basierten Tests zur Identifizierung von Genotypen in menschlichen Populationen begleitet.
Solche Studien sollten bei der Bewertung der Rolle von Chemikalien bei den vielen Umweltkrankheiten gegenwärtig unbekannter Herkunft besonders aufschlussreich sein. Die Berücksichtigung mehrerer arzneimittelmetabolisierender Enzympolymorphismen in Kombination (z. B. Tabelle 1) dürfte ebenfalls ein besonders fruchtbares Forschungsgebiet darstellen. Solche Studien werden die Rolle von Chemikalien bei der Verursachung von Krebs klären. Insgesamt sollten diese Informationen die Formulierung zunehmend individueller Ratschläge zur Vermeidung von Chemikalien ermöglichen, die wahrscheinlich von individueller Bedeutung sind. Dies ist das Gebiet der präventiven Toxikologie. Eine solche Beratung wird zweifellos allen Menschen bei der Bewältigung der ständig zunehmenden chemischen Belastung, der wir ausgesetzt sind, sehr helfen.