- You are here:
-
Home

-
Contents
-
Part IV. Tools and Approaches
-
Toxicology
- General Principles of Toxicology

General Principles of Toxicology
Definitions and Concepts
Exposure, Dose and Response
Toxicity is the intrinsic capacity of a chemical agent to affect an organism adversely.
Xenobiotics is a term for “foreign substances”, that is, foreign to the organism. Its opposite is endogenous compounds. Xenobiotics include drugs, industrial chemicals, naturally occurring poisons and environmental pollutants.
Hazard is the potential for the toxicity to be realized in a specific setting or situation.
Risk is the probability of a specific adverse effect to occur. It is often expressed as the percentage of cases in a given population and during a specific time period. A risk estimate can be based upon actual cases or a projection of future cases, based upon extrapolations.
Toxicity rating and toxicity classification can be used for regulatory purposes. Toxicity rating is an arbitrary grading of doses or exposure levels causing toxic effects. The grading can be “supertoxic,” “highly toxic,” “moderately toxic” and so on. The most common ratings concern acute toxicity. Toxicity classification concerns the grouping of chemicals into general categories according to their most important toxic effect. Such categories can include allergenic, neurotoxic, carcinogenic and so on. This classification can be of administrative value as a warning and as information.
The dose-effect relationship is the relationship between dose and effect on the individual level. An increase in dose may in- crease the intensity of an effect, or a more severe effect may result. A dose-effect curve may be obtained at the level of the whole organism, the cell or the target molecule. Some toxic effects, such as death or cancer, are not graded but are “all or none” effects.
The dose-response relationship is the relationship between dose and the percentage of individuals showing a specific effect. With increasing dose a greater number of individuals in the exposed population will usually be affected.
It is essential to toxicology to establish dose-effect and dose- response relationships. In medical (epidemiological) studies a criterion often used for accepting a causal relationship between an agent and a disease is that effect or response is proportional to dose.
Several dose-response curves can be drawn for a chemical—one for each type of effect. The dose-response curve for most toxic effects (when studied in large populations) has a sigmoid shape. There is usually a low-dose range where there is no response detected; as dose increases, the response follows an ascending curve that will usually reach a plateau at a 100% response. The dose-response curve reflects the variations among individuals in a population. The slope of the curve varies from chemical to chemical and between different types of effects. For some chemicals with specific effects (carcinogens, initiators, mutagens) the dose-response curve might be linear from dose zero within a certain dose range. This means that no threshold exists and that even small doses represent a risk. Above that dose range, the risk may increase at greater than a linear rate.
Variation in exposure during the day and the total length of exposure during one’s lifetime may be as important for the outcome (response) as mean or average or even integrated dose level. High peak exposures may be more harmful than a more even exposure level. This is the case for some organic solvents. On the other hand, for some carcinogens, it has been experimentally shown that the fractionation of a single dose into several exposures with the same total dose may be more effective in producing tumours.
A dose is often expressed as the amount of a xenobiotic entering an organism (in units such as mg/kg body weight). The dose may be expressed in different (more or less informative) ways: exposure dose, which is the air concentration of pollutant inhaled during a certain time period (in work hygiene usually eight hours), or the retained or absorbed dose (in industrial hygiene also called the body burden), which is the amount present in the body at a certain time during or after exposure. The tissue dose is the amount of substance in a specific tissue and the target dose is the amount of substance (usually a metabolite) bound to the critical molecule. The target dose can be expressed as mg chemical bound per mg of a specific macromolecule in the tissue. To apply this concept, information on the mechanism of toxic action on the molecular level is needed. The target dose is more exactly associated with the toxic effect. The exposure dose or body burden may be more easily available, but these are less precisely related to the effect.
In the dose concept a time aspect is often included, even if it is not always expressed. The theoretical dose according to Haber’s law is D = ct, where D is dose, c is concentration of the xenobiotic in the air and t the duration of exposure to the chemical. If this concept is used at the target organ or molecular level, the amount per mg tissue or molecule over a certain time may be used. The time aspect is usually more important for understanding repeated exposures and chronic effects than for single exposures and acute effects.
Additive effects occur as a result of exposure to a combination of chemicals, where the individual toxicities are simply added to each other (1+1= 2). When chemicals act via the same mechanism, additivity of their effects is assumed although not always the case in reality. Interaction between chemicals may result in an inhibition (antagonism), with a smaller effect than that expected from addition of the effects of the individual chemicals (1+1 2). Alternatively, a combination of chemicals may produce a more pronounced effect than would be expected by addition (increased response among individuals or an increase in frequency of response in a population), this is called synergism (1+1 >2).
Latency time is the time between first exposure and the appearance of a detectable effect or response. The term is often used for carcinogenic effects, where tumours may appear a long time after the start of exposure and sometimes long after the cessation of exposure.
A dose threshold is a dose level below which no observable effect occurs. Thresholds are thought to exist for certain effects, like acute toxic effects; but not for others, like carcinogenic effects (by DNA-adduct-forming initiators). The mere absence of a response in a given population should not, however, be taken as evidence for the existence of a threshold. Absence of response could be due to simple statistical phenomena: an adverse effect occurring at low frequency may not be detectable in a small population.
LD50 (effective dose) is the dose causing 50% lethality in an animal population. The LD50 is often given in older literature as a measure of acute toxicity of chemicals. The higher the LD50, the lower is the acute toxicity. A highly toxic chemical (with a low LD50) is said to be potent. There is no necessary correlation between acute and chronic toxicity. ED50 (effective dose) is the dose causing a specific effect other than lethality in 50% of the animals.
NOEL (NOAEL) means the no observed (adverse) effect level, or the highest dose that does not cause a toxic effect. To establish a NOEL requires multiple doses, a large population and additional information to make sure that absence of a response is not merely a statistical phenomenon. LOEL is the lowest observed effective dose on a dose-response curve, or the lowest dose that causes an effect.
A safety factor is a formal, arbitrary number with which one divides the NOEL or LOEL derived from animal experiments to obtain a tentative permissible dose for humans. This is often used in the area of food toxicology, but may be used also in occupational toxicology. A safety factor may also be used for extrapolation of data from small populations to larger populations. Safety factors range from 100 to 103. A safety factor of two may typically be sufficient to protect from a less serious effect (such as irritation) and a factor as large as 1,000 may be used for very serious effects (such as cancer). The term safety factor could be better replaced by the term protection factor or, even, uncertainty factor. The use of the latter term reflects scientific uncertainties, such as whether exact dose-response data can be translated from animals to humans for the particular chemical, toxic effect or exposure situation.
Extrapolations are theoretical qualitative or quantitative estimates of toxicity (risk extrapolations) derived from translation of data from one species to another or from one set of dose-response data (typically in the high dose range) to regions of dose-response where no data exist. Extrapolations usually must be made to predict toxic responses outside the observation range. Mathematical modelling is used for extrapolations based upon an understanding of the behaviour of the chemical in the organism (toxicokinetic modelling) or based upon the understanding of statistical probabilities that specific biological events will occur (biologically or mechanistically based models). Some national agencies have developed sophisticated extrapolation models as a formalized method to predict risks for regulatory purposes. (See discussion of risk assessment later in the chapter.)
Systemic effects are toxic effects in tissues distant from the route of absorption.
Target organ is the primary or most sensitive organ affected after exposure. The same chemical entering the body by different routes of exposure dose, dose rate, sex and species may affect different target organs. Interaction between chemicals, or between chemicals and other factors may affect different target organs as well.
Acute effects occur after limited exposure and shortly (hours, days) after exposure and may be reversible or irreversible.
Chronic effects occur after prolonged exposure (months, years, decades) and/or persist after exposure has ceased.
Acute exposure is an exposure of short duration, while chronic exposure is long-term (sometimes life-long) exposure.
Tolerance to a chemical may occur when repeat exposures result in a lower response than what would have been expected without pretreatment.
Uptake and Disposition
Transport processes
Diffusion. In order to enter the organism and reach a site where damage is produced, a foreign substance has to pass several barriers, including cells and their membranes. Most toxic substances pass through membranes passively by diffusion. This may occur for small water-soluble molecules by passage through aqueous channels or, for fat-soluble ones, by dissolution into and diffusion through the lipid part of the membrane. Ethanol, a small molecule that is both water and fat soluble, diffuses rapidly through cell membranes.
Diffusion of weak acids and bases. Weak acids and bases may readily pass membranes in their non-ionized, fat-soluble form while ionized forms are too polar to pass. The degree of ionization of these substances depends on pH. If a pH gradient exists across a membrane they will therefore accumulate on one side. The urinary excretion of weak acids and bases is highly dependent on urinary pH. Foetal or embryonic pH is somewhat higher than maternal pH, causing a slight accumulation of weak acids in the foetus or embryo.
Facilitated diffusion. The passage of a substance may be facilitated by carriers in the membrane. Facilitated diffusion is similar to enzyme processes in that it is protein mediated, highly selective, and saturable. Other substances may inhibit the facilitated transport of xenobiotics.
Active transport. Some substances are actively transported across cell membranes. This transport is mediated by carrier proteins in a process analogous to that of enzymes. Active transport is similar to facilitated diffusion, but it may occur against a concentration gradient. It requires energy input and a metabolic inhibitor can block the process. Most environmental pollutants are not transported actively. One exception is the active tubular secretion and reabsorption of acid metabolites in the kidneys.
Phagocytosis is a process where specialized cells such as macrophages engulf particles for subsequent digestion. This transport process is important, for example, for the removal of particles in the alveoli.
Bulk flow. Substances are also transported in the body along with the movement of air in the respiratory system during breathing, and the movements of blood, lymph or urine.
Filtration. Due to hydrostatic or osmotic pressure water flows in bulk through pores in the endothelium. Any solute that is small enough will be filtered together with the water. Filtration occurs to some extent in the capillary bed in all tissues but is particularly important in the formation of primary urine in the kidney glomeruli.
Absorption
Absorption is the uptake of a substance from the environment into the organism. The term usually includes not only the entrance into the barrier tissue but also the further transport into circulating blood.
Pulmonary absorption. The lungs are the primary route of deposition and absorption of small airborne particles, gases, vapours and aerosols. For highly water-soluble gases and vapours a significant part of the uptake occurs in the nose and the respiratory tree, but for less soluble substances it primarily takes place in the lung alveoli. The alveoli have a very large surface area (about 100m2 in humans). In addition, the diffusion barrier is extremely small, with only two thin cell layers and a distance in the order of micrometers from alveolar air to systemic blood circulation. This makes the lungs very efficient not only in the exchange of oxygen and carbon dioxide but also of other gases and vapours. In general, the diffusion across the alveolar wall is so rapid that it does not limit the uptake. The absorption rate is instead dependent on flow (pulmonary ventilation, cardiac output) and solubility (blood: air partition coefficient). Another important factor is metabolic elimination. The relative importance of these factors for pulmonary absorption varies greatly for different substances. Physical activity results in increased pulmonary ventilation and cardiac output, and decreased liver blood flow (and, hence, biotransformation rate). For many inhaled substances this leads to a marked increase in pulmonary absorption.
Percutaneous absorption. The skin is a very efficient barrier. Apart from its thermoregulatory role, it is designed to protect the organism from micro-organisms, ultraviolet radiation and other deleterious agents, and also against excessive water loss. The diffusion distance in the dermis is on the order of tenths of millimetres. In addition, the keratin layer has a very high resistance to diffusion for most substances. Nevertheless, significant dermal absorption resulting in toxicity may occur for some substances—highly toxic, fat-soluble substances such as organophosphorous insecticides and organic solvents, for example. Significant absorption is likely to occur after exposure to liquid substances. Percutaneous absorption of vapour may be important for solvents with very low vapour pressure and high affinity to water and skin.
Gastrointestinal absorption occurs after accidental or intentional ingestion. Larger particles originally inhaled and deposited in the respiratory tract may be swallowed after mucociliary transport to the pharynx. Practically all soluble substances are efficiently absorbed in the gastrointestinal tract. The low pH of the gut may facilitate absorption, for instance, of metals.
Other routes. In toxicity testing and other experiments, special routes of administration are often used for convenience, although these are rare and usually not relevant in the occupational setting. These routes include intravenous (IV), subcutaneous (sc), intraperitoneal (ip) and intramuscular (im) injections. In general, substances are absorbed at a higher rate and more completely by these routes, especially after IV injection. This leads to short-lasting but high concentration peaks that may increase the toxicity of a dose.
Distribution
The distribution of a substance within the organism is a dynamic process which depends on uptake and elimination rates, as well as the blood flow to the different tissues and their affinities for the substance. Water-soluble, small, uncharged molecules, univalent cations, and most anions diffuse easily and will eventually reach a relatively even distribution in the body.
Volume of distribution is the amount of a substance in the body at a given time, divided by the concentration in blood, plasma or serum at that time. The value has no meaning as a physical volume, as many substances are not uniformly distributed in the organism. A volume of distribution of less than one l/kg body weight indicates preferential distribution in the blood (or serum or plasma), whereas a value above one indicates a preference for peripheral tissues such as adipose tissue for fat soluble substances.
Accumulation is the build-up of a substance in a tissue or organ to higher levels than in blood or plasma. It may also refer to a gradual build-up over time in the organism. Many xenobiotics are highly fat soluble and tend to accumulate in adipose tissue, while others have a special affinity for bone. For example, calcium in bone may be exchanged for cations of lead, strontium, barium and radium, and hydroxyl groups in bone may be exchanged for fluoride.
Barriers. The blood vessels in the brain, testes and placenta have special anatomical features that inhibit passage of large molecules like proteins. These features, often referred to as blood-brain, blood-testes, and blood-placenta barriers, may give the false impression that they prevent passage of any substance. These barriers are of little or no importance for xenobiotics that can diffuse through cell membranes.
Blood binding. Substances may be bound to red blood cells or plasma components, or occur unbound in blood. Carbon monoxide, arsenic, organic mercury and hexavalent chromium have a high affinity for red blood cells, while inorganic mercury and trivalent chromium show a preference for plasma proteins. A number of other substances also bind to plasma proteins. Only the unbound fraction is available for filtration or diffusion into eliminating organs. Blood binding may therefore increase the residence time in the organism but decrease uptake by target organs.
Elimination
Elimination is the disappearance of a substance in the body. Elimination may involve excretion from the body or transformation to other substances not captured by a specific method of measurement. The rate of disappearance may be expressed by the elimination rate constant, biological half-time or clearance.
Concentration-time curve. The curve of concentration in blood (or plasma) versus time is a convenient way of describing uptake and disposition of a xenobiotic.
Area under the curve (AUC) is the integral of concentration in blood (plasma) over time. When metabolic saturation and other non-linear processes are absent, AUC is proportional to the absorbed amount of substance.
Biological half-time (or half-life) is the time needed after the end of exposure to reduce the amount in the organism to one-half. As it is often difficult to assess the total amount of a substance, measurements such as the concentration in blood (plasma) are used. The half-time should be used with caution, as it may change, for example, with dose and length of exposure. In addition, many substances have complex decay curves with several half-times.
Bioavailability is the fraction of an administered dose entering the systemic circulation. In the absence of presystemic clearance, or first-pass metabolism, the fraction is one. In oral exposure presystemic clearance may be due to metabolism within the gastrointestinal content, gut wall or liver. First-pass metabolism will reduce the systemic absorption of the substance and instead increase the absorption of metabolites. This may lead to a different toxicity pattern.
Clearance is the volume of blood (plasma) per unit time completely cleared of a substance. To distinguish from renal clearance, for example, the prefix total, metabolic or blood (plasma) is often added.
Intrinsic clearance is the capacity of endogenous enzymes to transform a substance, and is also expressed in volume per unit time. If the intrinsic clearance in an organ is much lower than the blood flow, the metabolism is said to be capacity limited. Conversely, if the intrinsic clearance is much higher than the blood flow, the metabolism is flow limited.
Excretion
Excretion is the exit of a substance and its biotransformation products from the organism.
Excretion in urine and bile. The kidneys are the most important excretory organs. Some substances, especially acids with high molecular weights, are excreted with bile. A fraction of biliary excreted substances may be reabsorbed in the intestines. This process, enterohepatic circulation, is common for conjugated substances following intestinal hydrolysis of the conjugate.
Other routes of excretion. Some substances, such as organic solvents and breakdown products such as acetone, are volatile enough so that a considerable fraction may be excreted by exhalation after inhalation. Small water-soluble molecules as well as fat-soluble ones are readily secreted to the foetus via the placenta, and into milk in mammals. For the mother, lactation can be a quantitatively important excretory pathway for persistent fat-soluble chemicals. The offspring may be secondarily exposed via the mother during pregnancy as well as during lactation. Water-soluble compounds may to some extent be excreted in sweat and saliva. These routes are generally of minor importance. However, as a large volume of saliva is produced and swallowed, saliva excretion may contribute to reabsorption of the compound. Some metals such as mercury are excreted by binding permanently to the sulphydryl groups of the keratin in the hair.
Toxicokinetic models
Mathematical models are important tools to understand and describe the uptake and disposition of foreign substances. Most models are compartmental, that is, the organism is represented by one or more compartments. A compartment is a chemically and physically theoretical volume in which the substance is assumed to distribute homogeneously and instantaneously. Simple models may be expressed as a sum of exponential terms, while more complicated ones require numerical procedures on a computer for their solution. Models may be subdivided in two categories, descriptive and physiological.
In descriptive models, fitting to measured data is performed by changing the numerical values of the model parameters or even the model structure itself. The model structure normally has little to do with the structure of the organism. Advantages of the descriptive approach are that few assumptions are made and that there is no need for additional data. A disadvantage of descriptive models is their limited usefulness for extrapolations.
Physiological models are constructed from physiological, anatomical and other independent data. The model is then refined and validated by comparison with experimental data. An advantage of physiological models is that they can be used for extrapolation purposes. For example, the influence of physical activity on the uptake and disposition of inhaled substances may be predicted from known physiological adjustments in ventilation and cardiac output. A disadvantage of physiological models is that they require a large amount of independent data.
Biotransformation
Biotransformation is a process which leads to a metabolic conversion of foreign compounds (xenobiotics) in the body. The process is often referred to as metabolism of xenobiotics. As a general rule metabolism converts lipid-soluble xenobiotics to large, water- soluble metabolites that can be effectively excreted.
The liver is the main site of biotransformation. All xenobiotics taken up from the intestine are transported to the liver by a single blood vessel (vena porta). If taken up in small quantities a foreign substance may be completely metabolized in the liver before reaching the general circulation and other organs (first pass effect). Inhaled xenobiotics are distributed via the general circulation to the liver. In that case only a fraction of the dose is metabolized in the liver before reaching other organs.
Liver cells contain several enzymes that oxidize xenobiotics. This oxidation generally activates the compound—it becomes more reactive than the parent molecule. In most cases the oxidized metabolite is further metabolized by other enzymes in a second phase. These enzymes conjugate the metabolite with an endogenous substrate, so that the molecule becomes larger and more polar. This facilitates excretion.
Enzymes that metabolize xenobiotics are also present in other organs such as the lungs and kidneys. In these organs they may play specific and qualitatively important roles in the metabolism of certain xenobiotics. Metabolites formed in one organ may be further metabolized in a second organ. Bacteria in the intestine may also participate in biotransformation.
Metabolites of xenobiotics can be excreted by the kidneys or via the bile. They can also be exhaled via the lungs, or bound to endogenous molecules in the body.
The relationship between biotransformation and toxicity is complex. Biotransformation can be seen as a necessary process for survival. It protects the organism against toxicity by preventing accumulation of harmful substances in the body. However, reactive intermediary metabolites may be formed in biotransformation, and these are potentially harmful. This is called metabolic activation. Thus, biotransformation may also induce toxicity. Oxidized, intermediary metabolites that are not conjugated can bind to and damage cellular structures. If, for example, a xenobiotic metabolite binds to DNA, a mutation can be induced (see “Genetic toxicology”). If the biotransformation system is overloaded, a massive destruction of essential proteins or lipid membranes may occur. This can result in cell death (see “Cellular injury and cellular death”).
Metabolism is a word often used interchangeably with biotransformation. It denotes chemical breakdown or synthesis reactions catalyzed by enzymes in the body. Nutrients from food, endogenous compounds, and xenobiotics are all metabolized in the body.
Metabolic activation means that a less reactive compound is converted to a more reactive molecule. This usually occurs during Phase 1 reactions.
Metabolic inactivation means that an active or toxic molecule is converted to a less active metabolite. This usually occurs during Phase 2 reactions. In certain cases an inactivated metabolite might be reactivated, for example by enzymatic cleavage.
Phase 1 reaction refers to the first step in xenobiotic metabolism. It usually means that the compound is oxidized. Oxidation usually makes the compound more water soluble and facilitates further reactions.
Cytochrome P450 enzymes are a group of enzymes that preferentially oxidize xenobiotics in Phase 1 reactions. The different enzymes are specialized for handling specific groups of xenobiotics with certain characteristics. Endogenous molecules are also substrates. Cytochrome P450 enzymes are induced by xenobiotics in a specific fashion. Obtaining induction data on cytochrome P450 can be informative about the nature of previous exposures (see “Genetic determinants of toxic response”).
Phase 2 reaction refers to the second step in xenobiotic meta- bolism. It usually means that the oxidized compound is conjugated with (coupled to) an endogenous molecule. This reaction increases the water solubility further. Many conjugated meta- bolites are actively excreted via the kidneys.
Transferases are a group of enzymes that catalyze Phase 2 reactions. They conjugate xenobiotics with endogenous compounds such as glutathione, amino acids, glucuronic acid or sulphate.
Glutathione is an endogenous molecule, a tripeptide, that is conjugated with xenobiotics in Phase 2 reactions. It is present in all cells (and in liver cells in high concentrations), and usually protects from activated xenobiotics. When glutathione is depleted, toxic reactions between activated xenobiotic metabolites and proteins, lipids or DNA may occur.
Induction means that enzymes involved in biotransformation are increased (in activity or amount) as a response to xenobiotic exposure. In some cases within a few days enzyme activity can be increased several fold. Induction is often balanced so that both Phase 1 and Phase 2 reactions are increased simultaneously. This may lead to a more rapid biotransformation and can explain tolerance. In contrast, unbalanced induction may increase toxicity.
Inhibition of biotransformation can occur if two xenobiotics are metabolized by the same enzyme. The two substrates have to compete, and usually one of the substrates is preferred. In that case the second substrate is not metabolized, or only slowly metabolized. As with induction, inhibition may increase as well as decrease toxicity.
Oxygen activation can be triggered by metabolites of certain xenobiotics. They may auto-oxidize under the production of activated oxygen species. These oxygen-derived species, which include superoxide, hydrogen peroxide and the hydroxyl radical, may damage DNA, lipids and proteins in cells. Oxygen activation is also involved in inflammatory processes.
Genetic variability between individuals is seen in many genes coding for Phase 1 and Phase 2 enzymes. Genetic variability may explain why certain individuals are more susceptible to toxic effects of xenobiotics than others.
Toxicokinetics
The human organism represents a complex biological system on various levels of organization, from the molecular-cellular level to the tissues and organs. The organism is an open system, exchanging matter and energy with the environment through numerous biochemical reactions in a dynamic equilibrium. The environment can be polluted, or contaminated with various toxicants.
Penetration of molecules or ions of toxicants from the work or living environment into such a strongly coordinated biological system can reversibly or irreversibly disturb normal cellular biochemical processes, or even injure and destroy the cell (see “Cellular injury and cellular death”).
Penetration of a toxicant from the environment to the sites of its toxic effect inside the organism can be divided into three phases:
- The exposure phase encompasses all processes occurring between various toxicants and/or the influence on them of environmental factors (light, temperature, humidity, etc.). Chemical transformations, degradation, biodegradation (by micro-organisms) as well as disintegration of toxicants can occur.
- The toxicokinetic phase encompasses absorption of toxicants into the organism and all processes which follow transport by body fluids, distribution and accumulation in tissues and organs, biotransformation to metabolites and elimination (excretion) of toxicants and/or metabolites from the organism.
- The toxicodynamic phase refers to the interaction of toxicants (molecules, ions, colloids) with specific sites of action on or in- side the cells—receptors—ultimately producing a toxic effect.
Here we will focus our attention exclusively on the toxicokinetic processes inside the human organism following exposure to toxicants in the environment.
The molecules or ions of toxicants present in the environment will penetrate into the organism through the skin and mucosa, or the epithelial cells of the respiratory and gastrointestinal tracts, depending on the point of entry. That means molecules and ions of toxicants must penetrate through cellular membranes of these biological systems, as well as through an intricate system of endomembranes inside the cell.
All toxicokinetic and toxicodynamic processes occur on the molecular-cellular level. Numerous factors influence these processes and these can be divided into two basic groups:
- chemical constitution and physicochemical properties of toxicants
- structure of the cell especially properties and function of membranes around the cell and its interior organelles.
Physico-Chemical Properties of Toxicants
In 1854 the Russian toxicologist E.V. Pelikan started studies on the relation between the chemical structure of a substance and its biological activity—the structure activity relationship (SAR). Chemical structure directly determines physico-chemical properties, some of which are responsible for biological activity.
To define the chemical structure numerous parameters can be selected as descriptors, which can be divided into various groups:
1. Physico-chemical:
- general—melting point, boiling point, vapour pressure, dissociation constant (pKa)
- electric—ionization potential, dielectric constant, dipole moment, mass: charge ratio, etc.
- quantum chemical—atomic charge, bond energy, resonance energy, electron density, molecular reactivity, etc.
2. Steric: molecular volume, shape and surface area, substructure shape, molecular reactivity, etc.
3. Structural: number of bonds number of rings (in polycyclic compounds), extent of branching, etc.
For each toxicant it is necessary to select a set of descriptors related to a particular mechanism of activity. However, from the toxicokinetic point of view two parameters are of general importance for all toxicants:
- The Nernst partition coefficient (P) establishes the solubility of toxicant molecules in the two-phase octanol (oil)-water system, correlating to their lipo- or hydrosolubility. This parameter will greatly influence the distribution and accumulation of toxicant molecules in the organism.
- The dissociation constant (pKa) defines the degree of ionization (electrolytic dissociation) of molecules of a toxicant into charged cations and anions at a particular pH. This constant represents the pH at which 50% ionization is achieved. Molecules can be lipophilic or hydrophilic, but ions are soluble exclusively in the water of body fluids and tissues. Knowing pKa it is possible to calculate the degree of ionization of a substance for each pH using the Henderson-Hasselbach equation.
For inhaled dusts and aerosols, the particle size, shape, surface area and density also influence their toxicokinetics and toxico- dynamics.
Structure and Properties of Membranes
The eukaryotic cell of human and animal organisms is encircled by a cytoplasmic membrane regulating the transport of substances and maintaining cell homeostasis. The cell organelles (nucleus, mitochondria) possess membranes too. The cell cytoplasm is compartmentalized by intricate membranous structures, the endo- plasmic reticulum and Golgi complex (endomembranes). All these membranes are structurally alike, but vary in the content of lipids and proteins.
The structural framework of membranes is a bilayer of lipid molecules (phospholipids, sphyngolipids, cholesterol). The backbone of a phospholipid molecule is glycerol with two of its -OH groups esterified by aliphatic fatty acids with 16 to 18 carbon atoms, and the third group esterified by a phosphate group and a nitrogenous compound (choline, ethanolamine, serine). In sphyngolipids, sphyngosine is the base.
The lipid molecule is amphipatic because it consists of a polar hydrophilic “head” (amino alcohol, phosphate, glycerol) and a non-polar twin “tail” (fatty acids). The lipid bilayer is arranged so that the hydrophilic heads constitute the outer and inner surface of membrane and lipophilic tails are stretched toward the membrane interior, which contains water, various ions and molecules.
Proteins and glycoproteins are inserted into the lipid bilayer (intrinsic proteins) or attached to the membrane surface (extrinsic proteins). These proteins contribute to the structural integrity of the membrane, but they may also perform as enzymes, carriers, pore walls or receptors.
The membrane represents a dynamic structure which can be disintegrated and rebuilt with a different proportion of lipids and proteins, according to functional needs.
Regulation of transport of substances into and out of the cell represents one of the basic functions of outer and inner membranes.
Some lipophilic molecules pass directly through the lipid bilayer. Hydrophilic molecules and ions are transported via pores. Membranes respond to changing conditions by opening or sealing certain pores of various sizes.
The following processes and mechanisms are involved in the transport of substances, including toxicants, through membranes:
- diffusion through lipid bilayer
- diffusion through pores
- transport by a carrier (facilitated diffusion).
Active processes:
- active transport by a carrier
- endocytosis (pinocytosis).
Diffusion
This represents the movement of molecules and ions through lipid bilayer or pores from a region of high concentration, or high electric potential, to a region of low concentration or potential (“downhill”). Difference in concentration or electric charge is the driving force influencing the intensity of the flux in both directions. In the equilibrium state, influx will be equal to efflux. The rate of diffusion follows Ficke’s law, stating that it is directly proportional to the available surface of membrane, difference in concentration (charge) gradient and characteristic diffusion coefficient, and inversely proportional to the membrane thickness.
Small lipophilic molecules pass easily through the lipid layer of membrane, according to the Nernst partition coefficient.
Large lipophilic molecules, water soluble molecules and ions will use aqueous pore channels for their passage. Size and stereoconfiguration will influence passage of molecules. For ions, besides size, the type of charge will be decisive. The protein molecules of pore walls can gain positive or negative charge. Narrow pores tend to be selective—negatively charged ligands will allow passage only for cations, and positively charged ligands will allow passage only for anions. With the increase of pore diameter hydrodynamic flow is dominant, allowing free passage of ions and molecules, according to Poiseuille’s law. This filtration is a consequence of the osmotic gradient. In some cases ions can penetrate through specific complex molecules—ionophores—which can be produced by micro-organisms with antibiotic effects (nonactin, valinomycin, gramacidin, etc.).
Facilitated or catalyzed diffusion
This requires the presence of a carrier in the membrane, usually a protein molecule (permease). The carrier selectively binds substances, resembling a substrate-enzyme complex. Similar molecules (including toxicants) can compete for the specific carrier until its saturation point is reached. Toxicants can compete for the carrier and when they are irreversibly bound to it the transport is blocked. The rate of transport is characteristic for each type of carrier. If transport is performed in both direction, it is called exchange diffusion.
Active transport
For transport of some substances vital for the cell, a special type of carrier is used, transporting against the concentration gradient or electric potential (“uphill”). The carrier is very stereospecific and can be saturated.
For uphill transport, energy is required. The necessary energy is obtained by catalytic cleavage of ATP molecules to ADP by the enzyme adenosine triphosphatase (ATP-ase).
Toxicants can interfere with this transport by competitive or non-competitive inhibition of the carrier or by inhibition of ATP-ase activity.
Endocytosis
Endocytosis is defined as a transport mechanism in which the cell membrane encircles material by enfolding to form a vesicle transporting it through the cell. When the material is liquid, the process is termed pinocytosis. In some cases the material is bound to a receptor and this complex is transported by a membrane vesicle. This type of transport is especially used by epithelial cells of the gastrointestinal tract, and cells of the liver and kidneys.
Absorption of Toxicants
People are exposed to numerous toxicants present in the work and living environment, which can penetrate into the human organism by three main portals of entry:
- via the respiratory tract by inhalation of polluted air
- via the gastrointestinal tract by ingestion of contaminated food, water and drinks
- through the skin by dermal, cutaneous penetration.
In the case of exposure in industry, inhalation represents the dominant way of entry of toxicants, followed by dermal penetration. In agriculture, pesticides exposure via dermal absorption is almost equal to cases of combined inhalation and dermal penetration. The general population is mostly exposed by ingestion of contaminated food, water and beverages, then by inhalation and less often by dermal penetration.
Absorption via the respiratory tract
Absorption in the lungs represents the main route of uptake for numerous airborne toxicants (gases, vapours, fumes, mists, smokes, dusts, aerosols, etc.).
The respiratory tract (RT) represents an ideal gas-exchange system possessing a membrane with a surface of 30m2 (expiration) to 100m2 (deep inspiration), behind which a network of about 2,000km of capillaries is located. The system, developed through evolution, is accommodated into a relatively small space (chest cavity) protected by ribs.
Anatomically and physiologically the RT can be divided into three compartments:
- the upper part of RT, or nasopharingeal (NP), starting at nose nares and extended to the pharynx and larynx; this part serves as an air-conditioning system
- the tracheo-bronchial tree (TB), encompassing numerous tubes of various sizes, which bring air to the lungs
- the pulmonary compartment (P), which consists of millions of alveoli (air-sacs) arranged in grapelike clusters.
Hydrophilic toxicants are easily absorbed by the epithelium of the nasopharingeal region. The whole epithelium of the NP and TB regions is covered by a film of water. Lipophilic toxicants are partially absorbed in the NP and TB, but mostly in the alveoli by diffusion through alveolo-capillary membranes. The absorption rate depends on lung ventilation, cardiac output (blood flow through lungs), solubility of toxicant in blood and its metabolic rate.
In the alveoli, gas exchange is carried out. The alveolar wall is made up of an epithelium, an interstitial framework of basement membrane, connective tissue and the capillary endothelium. The diffusion of toxicants is very rapid through these layers, which have a thickness of about 0.8 μm. In alveoli, toxicant is transferred from the air phase into the liquid phase (blood). The rate of absorption (air to blood distribution) of a toxicant depends on its concentration in alveolar air and the Nernst partition coefficient for blood (solubility coefficient).
In the blood the toxicant can be dissolved in the liquid phase by simple physical processes or bound to the blood cells and/or plasma constituents according to chemical affinity or by adsorption. The water content of blood is 75% and, therefore, hydrophilic gases and vapours show a high solubility in plasma (e.g., alcohols). Lipophilic toxicants (e.g., benzene) are usually bound to cells or macro-molecules such as albumen.
From the very beginning of exposure in the lungs, two opposite processes are occurring: absorption and desorption. The equilibrium between these processes depends on the concentration of toxicant in alveolar air and blood. At the onset of exposure the toxicant concentration in the blood is 0 and retention in blood is almost 100%. With continuation of exposure, an equilibrium between absorption and desorption is attained. Hydrophilic toxicants will rapidly attain equilibrium, and the rate of absorption depends on pulmonary ventilation rather than on blood flow. Lipophilic toxicants need a longer time to achieve equilibrium, and here the flow of unsaturated blood governs the rate of absorption.
Deposition of particles and aerosols in the RT depends on physical and physiological factors, as well as particle size. In short, the smaller the particle the deeper it will penetrate into the RT.
Relatively constant low retention of dust particles in the lungs of persons who are highly exposed (e.g., miners) suggests the existence of a very efficient system for the clearance of particles. In the upper part of the RT (tracheo-bronchial) a mucociliary blanket performs the clearance. In the pulmonary part, three different mechanisms are at work.: (1) mucociliary blanket, (2) phagocytosis and (3) direct penetration of particles through the alveolar wall.
The first 17 of the 23 branchings of the tracheo-bronchial tree possess ciliated epithelial cells. By their strokes these cilia constantly move a mucous blanket toward the mouth. Particles deposited on this mucociliary blanket will be swallowed in the mouth (ingestion). A mucous blanket also covers the surface of the alveolar epithelium, moving toward the mucociliary blanket. Additionally the specialized moving cells—phagocytes—engulf particles and micro-organisms in the alveoli and migrate in two possible directions:
- toward the mucociliary blanket, which transports them to the mouth
- through the intercellular spaces of the alveolar wall to the lymphatic system of the lungs; also particles can directly penetrate by this route.
Absorption via gastrointestinal tract
Toxicants can be ingested in the case of accidental swallowing, intake of contaminated food and drinks, or swallowing of particles cleared from the RT.
The entire alimentary channel, from oesophagus to anus, is basically built in the same way. A mucous layer (epithelium) is supported by connective tissue and then by a network of capillaries and smooth muscle. The surface epithelium of the stomach is very wrinkled to increase the absorption/secretion surface area. The intestinal area contains numerous small projections (villi), which are able to absorb material by “pumping in”. The active area for absorption in the intestines is about 100m2.
In the gastrointestinal tract (GIT) all absorption processes are very active:
- transcellular transport by diffusion through the lipid layer and/or pores of cell membranes, as well as pore filtration
- paracellular diffusion through junctions between cells
- facilitated diffusion and active transport
- endocytosis and the pumping mechanism of the villi.
Some toxic metal ions use specialized transport systems for essential elements: thallium, cobalt and manganese use the iron system, while lead appears to use the calcium system.
Many factors influence the rate of absorption of toxicants in various parts of the GIT:
- physico-chemical properties of toxicants, especially the Nernst partition coefficient and the dissociation constant; for particles, particle size is important—the smaller the size, the higher the solubility
- quantity of food present in the GIT (diluting effect)
- residence time in each part of the GIT (from a few minutes in the mouth to one hour in the stomach to many hours in the intestines
- the absorption area and absorption capacity of the epithelium
- local pH, which governs absorption of dissociated toxicants; in the acid pH of the stomach, non-dissociated acidic compounds will be more quickly absorbed
- peristalsis (movement of intestines by muscles) and local blood flow
- gastric and intestinal secretions transform toxicants into more or less soluble products; bile is an emulsifying agent producing more soluble complexes (hydrotrophy)
- combined exposure to other toxicants, which can produce synergistic or antagonistic effects in absorption processes
- presence of complexing/chelating agents
- the action of microflora of the RT (about 1.5kg), about 60 different bacterial species which can perform biotransformation of toxicants.
It is also necessary to mention the enterohepatic circulation. Polar toxicants and/or metabolites (glucuronides and other conjugates) are excreted with the bile into the duodenum. Here the enzymes of the microflora perform hydrolysis and liberated products can be reabsorbed and transported by the portal vein into the liver. This mechanism is very dangerous in the case of hepatotoxic substances, enabling their temporary accumulation in the liver.
In the case of toxicants biotransformed in the liver to less toxic or non-toxic metabolites, ingestion may represent a less dangerous portal of entry. After absorption in the GIT these toxicants will be transported by the portal vein to the liver, and there they can be partially detoxified by biotransformation.
Absorption through the skin (dermal, percutaneous)
The skin (1.8 m2 of surface in a human adult) together with the mucous membranes of the body orifices, covers the surface of the body. It represents a barrier against physical, chemical and biological agents, maintaining the body integrity and homeostasis and performing many other physiological tasks.
Basically the skin consists of three layers: epidermis, true skin (dermis) and subcutaneous tissue (hypodermis). From the toxicological point of view the epidermis is of most interest here. It is built of many layers of cells. A horny surface of flattened, dead cells (stratum corneum) is the top layer, under which a continuous layer of living cells (stratum corneum compactum) is located, followed by a typical lipid membrane, and then by stratum lucidum, stratum gramulosum and stratum mucosum. The lipid membrane represents a protective barrier, but in hairy parts of the skin, both hair follicles and sweat gland channels penetrate through it. Therefore, dermal absorption can occur by the following mechanisms:
- transepidermal absorption by diffusion through the lipid membrane (barrier), mostly by lipophilic substances (organic solvents, pesticides, etc.) and to a small extent by some hydrophilic substances through pores
- transfollicular absorption around the hair stalk into the hair follicle, bypassing the membrane barrier; this absorption occurs only in hairy areas of skin
- absorption via the ducts of sweat glands, which have a cross-sectional area of about 0.1to 1% of the total skin area (relative absorption is in this proportion)
- absorption through skin when injured mechanically, thermally, chemically or by skin diseases; here the skin layers, including lipid barrier, are disrupted and the way is open for toxicants and harmful agents to enter.
The rate of absorption through the skin will depend on many factors:
- concentration of toxicant, type of vehicle (medium), presence of other substances
- water content of skin, pH, temperature, local blood flow, perspiration, surface area of contaminated skin, thickness of skin
- anatomical and physiological characteristics of the skin due to sex, age, individual variations, differences occurring in various ethnic groups and races, etc.
Transport of Toxicants by Blood and Lymph
After absorption by any of these portals of entry, toxicants will reach the blood, lymph or other body fluids. The blood represents the major vehicle for transport of toxicants and their metabolites.
Blood is a fluid circulating organ, transporting necessary oxygen and vital substances to the cells and removing waste products of metabolism. Blood also contains cellular components, hormones, and other molecules involved in many physiological functions. Blood flows inside a relatively well closed, high-pressure circulatory system of blood vessels, pushed by the activity of the heart. Due to high pressure, leakage of fluid occurs. The lymphatic system represents the drainage system, in the form of a fine mesh of small, thin-walled lymph capillaries branching through the soft tissues and organs.
Blood is a mixture of a liquid phase (plasma, 55%) and solid blood cells (45%). Plasma contains proteins (albumins, globulins, fibrinogen), organic acids (lactic, glutamic, citric) and many other substances (lipids, lipoproteins, glycoproteins, enzymes, salts, xenobiotics, etc.). Blood cell elements include erythrocytes (Er), leukocytes, reticulocytes, monocytes, and platelets.
Toxicants are absorbed as molecules and ions. Some toxicants at blood pH form colloid particles as a third form in this liquid. Molecules, ions and colloids of toxicants have various possibilities for transport in blood:
- to be physically or chemically bound to the blood elements, mostly Er
- to be physically dissolved in plasma in a free state
- to be bound to one or more types of plasma proteins, complexed with the organic acids or attached to other fractions of plasma.
Most of the toxicants in blood exist partially in a free state in plasma and partially bound to erythrocytes and plasma constituents. The distribution depends on the affinity of toxicants to these constituents. All fractions are in a dynamic equilibrium.
Some toxicants are transported by the blood elements—mostly by erythrocytes, very rarely by leukocytes. Toxicants can be adsorbed on the surface of Er, or can bind to the ligands of stroma. If they penetrate into Er they can bind to the haem (e.g. carbon monoxide and selenium) or to the globin (Sb111, Po210). Some toxicants transported by Er are arsenic, cesium, thorium, radon, lead and sodium. Hexavalent chromium is exclusively bound to the Er and trivalent chromium to the proteins of plasma. For zinc, competition between Er and plasma occurs. About 96% of lead is transported by Er. Organic mercury is mostly bound to Er and inorganic mercury is carried mostly by plasma albumin. Small fractions of beryllium, copper, tellurium and uranium are carried by Er.
The majority of toxicants are transported by plasma or plasma proteins. Many electrolytes are present as ions in an equilibrium with non-dissociated molecules free or bound to the plasma fractions. This ionic fraction of toxicants is very diffusible, penetrating through the walls of capillaries into tissues and organs. Gases and vapours can be dissolved in the plasma.
Plasma proteins possess a total surface area of about 600to 800km2 offered for absorption of toxicants. Albumin molecules possess about 109 cationic and 120 anionic ligands at the disposal of ions. Many ions are partially carried by albumin (e.g., copper, zinc and cadmium), as are such compounds as dinitro- and ortho-cresols, nitro- and halogenated derivatives of aromatic hydrocarbons, and phenols.
Globulin molecules (alpha and beta) transport small molecules of toxicants as well as some metallic ions (copper, zinc and iron) and colloid particles. Fibrinogen shows affinity for certain small molecules. Many types of bonds can be involved in binding of toxicants to plasma proteins: Van der Waals forces, attraction of charges, association between polar and non-polar groups, hydrogen bridges, covalent bonds.
Plasma lipoproteins transport lipophilic toxicants such as PCBs. The other plasma fractions serve as a transport vehicle too. The affinity of toxicants for plasma proteins suggests their affinity for proteins in tissues and organs during distribution.
Organic acids (lactic, glutaminic, citric) form complexes with some toxicants. Alkaline earths and rare earths, as well as some heavy elements in the form of cations, are complexed also with organic oxy- and amino acids. All these complexes are usually diffusible and easily distributed in tissues and organs.
Physiologically chelating agents in plasma such as transferrin and metallothionein compete with organic acids and amino acids for cations to form stable chelates.
Diffusible free ions, some complexes and some free molecules are easily cleared from the blood into tissues and organs. The free fraction of ions and molecules is in a dynamic equilibrium with the bound fraction. The concentration of a toxicant in blood will govern the rate of its distribution into tissues and organs, or its mobilization from them into the blood.
Distribution of Toxicants in the Organism
The human organism can be divided into the following compartments. (1) internal organs, (2) skin and muscles, (3) adipose tissues, (4) connective tissue and bones. This classification is mostly based on the degree of vascular (blood) perfusion in a decreasing order. For example internal organs (including the brain), which represent only 12% of the total body weight, receive about 75% of the total blood volume. On the other hand, connective tissues and bones (15% of total body weight) receive only one per cent of the total blood volume.
The well-perfused internal organs generally achieve the highest concentration of toxicants in the shortest time, as well as an equilibrium between blood and this compartment. The uptake of toxicants by less perfused tissues is much slower, but retention is higher and duration of stay much longer (accumulation) due to low perfusion.
Three components are of major importance for the intracellular distribution of toxicants: content of water, lipids and proteins in the cells of various tissues and organs. The above-mentioned order of compartments also follows closely a decreasing water content in their cells. Hydrophilic toxicants will be more rapidly distributed to the body fluids and cells with high water content, and lipophilic toxicants to cells with higher lipid content (fatty tissue).
The organism possesses some barriers which impair penetration of some groups of toxicants, mostly hydrophilic, to certain organs and tissues, such as:
- the blood-brain barrier (cerebrospinal barrier), which restricts penetration of large molecules and hydrophilic toxicants to the brain and CNS; this barrier consists of a closely joined layer of endothelial cells; thus, lipophilic toxicants can penetrate through it
- the placental barrier, which has a similar effect on penetration of toxicants into the foetus from the blood of the mother
- the histo-haematologic barrier in the walls of capillaries, which is permeable for small- and intermediate-sized molecules, and for some larger molecules, as well as ions.
As previously noted only the free forms of toxicants in plasma (molecules, ions, colloids) are available for penetration through the capillary walls participating in distribution. This free fraction is in a dynamic equilibrium with the bound fraction. Concentration of toxicants in blood is in a dynamic equilibrium with their concentration in organs and tissues, governing retention (accumulation) or mobilization from them.
The condition of the organism, functional state of organs (especially neuro-humoral regulation), hormonal balance and other factors play a role in distribution.
Retention of toxicant in a particular compartment is generally temporary and redistribution into other tissues can occur. Retention and accumulation is based on the difference between the rates of absorption and elimination. The duration of retention in a compartment is expressed by the biological half-life. This is the time interval in which 50% of the toxicant is cleared from the tissue or organ and redistributed, translocated or eliminated from the organism.
Biotransformation processes occur during distribution and retention in various organs and tissues. Biotransformation produces more polar, more hydrophilic metabolites, which are more easily eliminated. A low rate of biotransformation of a lipophilic toxicant will generally cause its accumulation in a compartment.
The toxicants can be divided into four main groups according to their affinity, predominant retention and accumulation in a particular compartment:
- Toxicants soluble in the body fluids are uniformly distributed according to the water content of compartments. Many monovalent cations (e.g., lithium, sodium, potassium, rubidium) and some anions (e.g., chlorine, bromine), are distributed according to this pattern.
- Lipophilic toxicants show a high affinity for lipid-rich organs (CNS) and tissues (fatty, adipose).
- Toxicants forming colloid particles are then trapped by specialized cells of the reticuloendothelial system (RES) of organs and tissues. Tri- and quadrivalent cations (lanthanum, cesium, hafnium) are distributed in the RES of tissues and organs.
- Toxicants showing a high affinity for bones and connective tissue (osteotropic elements, bone seekers) include divalent cations (e.g., calcium, barium, strontium, radon, beryllium, aluminium, cadmium, lead).
Accumulation in lipid-rich tissues
The “standard man” of 70kg body weight contains about 15% of body weight in the form of adipose tissue, increasing with obesity to 50%. However, this lipid fraction is not uniformly distributed. The brain (CNS) is a lipid-rich organ, and peripheral nerves are wrapped with a lipid-rich myelin sheath and Schwann cells. All these tissues offer possibilities for accumulation of lipophilic toxicants.
Numerous non-electrolytes and non-polar toxicants with a suitable Nernst partition coefficient will be distributed to this compartment, as well as numerous organic solvents (alcohols, aldehydes, ketones, etc.), chlorinated hydrocarbons (including organochlorine insecticides such as DDT), some inert gases (radon), etc.
Adipose tissue will accumulate toxicants due to its low vascularization and lower rate of biotransformation. Here accumulation of toxicants may represent a kind of temporary “neutralization” because of lack of targets for toxic effect. However, potential danger for the organism is always present due to the possibility of mobilization of toxicants from this compartment back to the circulation.
Deposition of toxicants in the brain (CNS) or lipid-rich tissue of the myelin sheath of the peripheral nervous system is very dangerous. The neurotoxicants are deposited here directly next to their targets. Toxicants retained in lipid-rich tissue of the endocrine glands can produce hormonal disturbances. Despite the blood-brain barrier, numerous neurotoxicants of a lipophilic nature reach the brain (CNS): anaesthetics, organic solvents, pesticides, tetraethyl lead, organomercurials, etc.
Retention in the reticuloendothelial system
In each tissue and organ a certain percentage of cells is specialized for phagocytic activity, engulfing micro-organisms, particles, colloid particles, and so on. This system is called the reticuloendothelial system (RES), comprising fixed cells as well as moving cells (phagocytes). These cells are present in non-active form. An increase of the above-mentioned microbes and particles will activate the cells up to a saturation point.
Toxicants in the form of colloids will be captured by the RES of organs and tissues. Distribution depends on the colloid particle size. For larger particles, retention in the liver will be favoured. With smaller colloid particles, more or less uniform distribution will occur between the spleen, bone marrow and liver. Clearance of colloids from the RES is very slow, although small particles are cleared relatively more quickly.
Accumulation in bones
About 60 elements can be identified as osteotropic elements, or bone seekers.
Osteotropic elements can be divided into three groups:
- Elements representing or replacing physiological constituents of the bone. Twenty such elements are present in higher quantities. The others appear in trace quantities. Under conditions of chronic exposure, toxic metals such as lead, aluminium and mercury can also enter the mineral matrix of bone cells.
- Alkaline earths and other elements forming cations with an ionic diameter similar to that of calcium are exchangeable with it in bone mineral. Also, some anions are exchangeable with anions (phosphate, hydroxyl) of bone mineral.
- Elements forming microcolloids (rare earths) may be adsorbed on the surface of bone mineral.
The skeleton of a standard man accounts for 10to 15% of the total body weight, representing a large potential storage depot for osteotropic toxicants. Bone is a highly specialized tissue consisting by volume of 54% minerals and 38% organic matrix. The mineral matrix of bone is hydroxyapatite, Ca10(PO4)6(OH)2 , in which the ratio of Ca to P is about 1.5 to one. The surface area of mineral available for adsorption is about 100m2 per g of bone.
Metabolic activity of the bones of the skeleton can be divided in two categories:
- active, metabolic bone, in which processes of resorption and new bone formation, or remodelling of existing bone, are very extensive
- stable bone with a low rate of remodelling or growth.
In the fetus, infant and young child metabolic bone (see “available skeleton”) represents almost 100% of the skeleton. With age this percentage of metabolic bone decreases. Incorporation of toxicants during exposure appears in the metabolic bone and in more slowly turning-over compartments.
Incorporation of toxicants into bone occurs in two ways:
- For ions, an ion exchange occurs with physiologically present calcium cations, or anions (phosphate, hydroxyl).
- For toxicants forming colloid particles, adsorption on the mineral surface occurs.
Ion-exchange reactions
The bone mineral, hydroxyapatite, represents a complex ion- exchange system. Calcium cations can be exchanged by various cations. The anions present in bone can also be exchanged by anions: phosphate with citrates and carbonates, hydroxyl with fluorine. Ions which are not exchangeable can be adsorbed on the mineral surface. When toxicant ions are incorporated in the mineral, a new layer of mineral can cover the mineral surface, burying toxicant into the bone structure. Ion exchange is a reversible process, depending on the concentration of ions, pH and fluid volume. Thus, for example, an increase of dietary calcium may decrease the deposition of toxicant ions in the lattice of minerals. It has been mentioned that with age the percentage of metabolic bone is decreased, although ion exchange continues. With ageing, bone mineral resorption occurs, in which bone density actually decreases. At this point, toxicants in bone may be released (e.g., lead).
About 30% of the ions incorporated into bone minerals are loosely bound and can be exchanged, captured by natural chelating agents and excreted, with a biological half-life of 15 days. The other 70% is more firmly bound. Mobilization and excretion of this fraction shows a biological half-life of 2.5 years and more depending on bone type (remodelling processes).
Chelating agents (Ca-EDTA, penicillamine, BAL, etc.) can mobilize considerable quantities of some heavy metals, and their excretion in urine greatly increased.
Colloid adsorption
Colloid particles are adsorbed as a film on the mineral surface (100m2 per g) by Van der Waals forces or chemisorption. This layer of colloids on the mineral surfaces is covered with the next layer of formed minerals, and the toxicants are more buried into the bone structure. The rate of mobilization and elimination depends on remodelling processes.
Accumulation in hair and nails
The hair and nails contain keratin, with sulphydryl groups able to chelate metallic cations such as mercury and lead.
Distribution of toxicant inside the cell
Recently the distribution of toxicants, especially some heavy metals, within cells of tissues and organs has become of importance. With ultracentrifugation techniques, various fractions of the cell can be separated to determine their content of metal ions and other toxicants.
Animal studies have revealed that after penetration into the cell, some metal ions are bound to a specific protein, metallothionein. This low molecular weight protein is present in the cells of liver, kidney and other organs and tissues. Its sulphydryl groups can bind six ions per molecule. Increased presence of metal ions induces the biosynthesis of this protein. Ions of cadmium are the most potent inducer. Metallothionein serves also to maintain homeostasis of vital copper and zinc ions. Metallothionein can bind zinc, copper, cadmium, mercury, bismuth, gold, cobalt and other cations.
Biotransformation and Elimination of Toxicants
During retention in cells of various tissues and organs, toxicants are exposed to enzymes which can biotransform (metabolize) them, producing metabolites. There are many pathways for the elimination of toxicants and/or metabolites: by exhaled air via the lungs, by urine via the kidneys, by bile via the GIT, by sweat via the skin, by saliva via the mouth mucosa, by milk via the mammary glands, and by hair and nails via normal growth and cell turnover.
The elimination of an absorbed toxicant depends on the portal of entry. In the lungs the absorption/desorption process starts immediately and toxicants are partially eliminated by exhaled air. Elimination of toxicants absorbed by other paths of entry is prolonged and starts after transport by blood, eventually being completed after distribution and biotransformation. During absorption an equilibrium exists between the concentrations of a toxicant in the blood and in tissues and organs. Excretion decreases toxicant blood concentration and may induce mobilization of a toxicant from tissues into blood.
Many factors can influence the elimination rate of toxicants and their metabolites from the body:
- physico-chemical properties of toxicants, especially the Nernst partition coefficient (P), dissociation constant (pKa), polarity, molecular structure, shape and weight
- level of exposure and time of post-exposure elimination
- portal of entry
- distribution in the body compartments, which differ in exchange rate with the blood and blood perfusion
- rate of biotransformation of lipophilic toxicants to more hydrophilic metabolites
- overall health condition of organism and, especially, of excretory organs (lungs, kidneys, GIT, skin, etc.)
- presence of other toxicants which can interfere with elimination.
Here we distinguish two groups of compartments: (1) the rapid-exchange system— in these compartments, tissue concentration of toxicant is similar to that of the blood; and (2) the slow-exchange system, where tissue concentration of toxicant is higher than in blood due to binding and accumulation—adipose tissue, skeleton and kidneys can temporarily retain some toxicants, e.g., arsenic and zinc.
A toxicant can be excreted simultaneously by two or more excretion routes. However, usually one route is dominant.
Scientists are developing mathematical models describing the excretion of a particular toxicant. These models are based on the movement from one or both compartments (exchange systems), biotransformation and so on.
Elimination by exhaled air via lungs
Elimination via the lungs (desorption) is typical for toxicants with high volatility (e.g., organic solvents). Gases and vapours with low solubility in blood will be quickly eliminated this way, whereas toxicants with high blood solubility will be eliminated by other routes.
Organic solvents absorbed by the GIT or skin are excreted partially by exhaled air in each passage of blood through the lungs, if they have a sufficient vapour pressure. The Breathalyser test used for suspected drunk drivers is based on this fact. The concentration of CO in exhaled air is in equilibrium with the CO-Hb blood content. The radioactive gas radon appears in exhaled air due to the decay of radium accumulated in the skeleton.
Elimination of a toxicant by exhaled air in relation to the post-exposure period of time usually is expressed by a three-phase curve. The first phase represents elimination of toxicant from the blood, showing a short half-life. The second, slower phase represents elimination due to exchange of blood with tissues and organs (quick-exchange system). The third, very slow phase is due to exchange of blood with fatty tissue and skeleton. If a toxicant is not accumulated in such compartments, the curve will be two-phase. In some cases a four-phase curve is also possible.
Determination of gases and vapours in exhaled air in the post-exposure period is sometimes used for evaluation of exposures in workers.
Renal excretion
The kidney is an organ specialized in the excretion of numerous water-soluble toxicants and metabolites, maintaining homeostasis of the organism. Each kidney possesses about one million nephrons able to perform excretion. Renal excretion represents a very complex event encompassing three different mechanisms:
- glomerular filtration by Bowman’s capsule
- active transport in the proximal tubule
- passive transport in the distal tubule.
Excretion of a toxicant via the kidneys to urine depends on the Nernst partition coefficient, dissociation constant and pH of urine, molecular size and shape, rate of metabolism to more hydrophilic metabolites, as well as health status of the kidneys.
The kinetics of renal excretion of a toxicant or its metabolite can be expressed by a two-, three- or four-phase excretion curve, depending on the distribution of the particular toxicant in various body compartments differing in the rate of exchange with the blood.
Saliva
Some drugs and metallic ions can be excreted through the mucosa of the mouth by saliva—for example, lead (“lead line”), mercury, arsenic, copper, as well as bromides, iodides, ethyl alcohol, alkaloids, and so on. The toxicants are then swallowed, reaching the GIT, where they can be reabsorbed or eliminated by faeces.
Sweat
Many non-electrolytes can be partially eliminated via skin by sweat: ethyl alcohol, acetone, phenols, carbon disulphide and chlorinated hydrocarbons.
Milk
Many metals, organic solvents and some organochlorine pesticides (DDT) are secreted via the mammary gland in mother’s milk. This pathway can represent a danger for nursing infants.
Hair
Analysis of hair can be used as an indicator of homeostasis of some physiological substances. Also exposure to some toxicants, especially heavy metals, can be evaluated by this kind of bioassay.
Elimination of toxicants from the body can be increased by:
- mechanical translocation via gastric lavage, blood transfusion or dialysis
- creating physiological conditions which mobilize toxicants by diet, change of hormonal balance, improving renal function by application of diuretics
- administration of complexing agents (citrates, oxalates, salicilates, phosphates), or chelating agents (Ca-EDTA, BAL, ATA, DMSA, penicillamine); this method is indicated only in persons under strict medical control. Application of chelating agents is often used for elimination of heavy metals from the body of exposed workers in the course of their medical treatment. This method is also used for evaluation of total body burden and level of past exposure.
Exposure Determinations
Determination of toxicants and metabolites in blood, exhaled air, urine, sweat, faeces and hair is more and more used for evaluation of human exposure (exposure tests) and/or evaluation of the degree of intoxication. Therefore biological exposure limits (Biological MAC Values, Biological Exposure Indices—BEI) have recently been established. These bioassays show “internal exposure” of the organism, that is, total exposure of the body in both the work and living environments by all portals of entry (see “Toxicology test methods: Biomarkers”).
Combined Effects Due to Multiple Exposure
People in the work and/or living environment are usually exposed simultaneously or consecutively to various physical and chemical agents. Also it is necessary to take into consideration that some persons use medications, smoke, consume alcohol and food containing additives and so on. That means that usually multiple exposure is occurring. Physical and chemical agents can interact in each step of toxicokinetic and/or toxicodynamic processes, producing three possible effects:
- Independent. Each agent produces a different effect due to a different mechanism of action,
- Synergistic. The combined effect is greater than that of each single agent. Here we differentiate two types: (a) additive, where the combined effect is equal to the sum of the effects produced by each agent separately and (b) potentiating, where the combined effect is greater than additive.
- Antagonistic. The combined effect is lower than additive.
However, studies on combined effects are rare. This kind of study is very complex due to the combination of various factors and agents.
We can conclude that when the human organism is exposed to two or more toxicants simultaneously or consecutively, it is necessary to consider the possibility of some combined effects, which can increase or decrease the rate of toxicokinetic processes.
Target Organ and Critical Effects
The priority objective of occupational and environmental toxicology is to improve the prevention or substantial limitation of health effects of exposure to hazardous agents in the general and occupational environments. To this end systems have been developed for quantitative risk assessment related to a given exposure (see the section “Regulatory toxicology”).
The effects of a chemical on particular systems and organs are related to the magnitude of exposure and whether exposure is acute or chronic. In view of the diversity of toxic effects even within one system or organ, a uniform philosophy concerning the critical organ and critical effect has been proposed for the purpose of risk assessment and development of health-based recommended concentration limits of toxic substances in different environmental media.
From the point of view of preventive medicine, it is of particular importance to identify early adverse effects, based on the general assumption that preventing or limiting early effects may prevent more severe health effects from developing.
Such an approach has been applied to heavy metals. Although heavy metals, such as lead, cadmium and mercury, belong to a specific group of toxic substances where the chronic effect of activity is dependent on their accumulation in the organs, the definitions presented below were published by the Task Group on Metal Toxicity (Nordberg 1976).
The definition of the critical organ as proposed by the Task Group on Metal Toxicity has been adopted with a slight modification: the word metal has been replaced with the expression potentially toxic substance (Duffus 1993).
Whether a given organ or system is regarded as critical depends not only on the toxicomechanics of the hazardous agent but also on the route of absorption and the exposed population.
- Critical concentration for a cell: the concentration at which adverse functional changes, reversible or irreversible, occur in the cell.
- Critical organ concentration: the mean concentration in the organ at the time at which the most sensitive type of cells in the organ reach critical concentration.
- Critical organ: that particular organ which first attains the critical concentration of metal under specified circumstances of exposure and for a given population.
- Critical effect: defined point in the relationship between dose and effect in the individual, namely the point at which an adverse effect occurs in cellular function of the critical organ. At an exposure level lower than that giving a critical concentration of metal in the critical organ, some effects may occur that do not impair cellular function per se, yet are detectable by means of biochemical and other tests. Such effects are defined as sub- critical effects.
The biological meaning of subcritical effect is sometimes not known; it may stand for exposure biomarker, adaptation index or a critical effect precursor (see “Toxicology test methods: Biomarkers”). The latter possibility can be particularly significant in view of prophylactic activities.
Table 1 displays examples of critical organs and effects for different chemicals. In chronic environmental exposure to cadmium, where the route of absorption is of minor importance (cadmium air concentrations range from 10 to 20μg/m3 in the urban and 1 to 2 μg/m3 in the rural areas), the critical organ is the kidney. In the occupational setting where the TLV reaches 50μg/m3 and inhalation constitutes the main route of exposure, two organs, lung and kidney, are regarded as critical.
Table 1. Examples of critical organs and critical effects
| Substance | Critical organ in chronic exposure | Critical effect |
| Cadmium | Lungs | Nonthreshold: Lung cancer (unit risk 4.6 x 10-3) |
| Kidney | Threshold: Increased excretion of low molecular proteins (β2 –M, RBP) in urine |
|
| Lungs | Emphysema slight function changes | |
| Lead | Adults Haematopoietic system |
Increased delta-aminolevulinic acid excretion in urine (ALA-U); increased concentration of free erythrocyte protoporphyrin (FEP) in erythrocytes |
| Peripheral nervous system | Slowing of the conduction velocities of the slower nerve fibres | |
| Mercury (elemental) | Young children Central nervous system |
Decrease in IQ and other subtle effects; mercurial tremor (fingers, lips, eyelids) |
| Mercury (mercuric) | Kidney | Proteinuria |
| Manganese | Adults Central nervous system |
Impairment of psychomotor functions |
| Children Lungs |
Respiratory symptoms | |
| Central nervous system | Impairment of psychomotor functions | |
| Toluene | Mucous membranes | Irritation |
| Vinyl chloride | Liver | Cancer (angiosarcoma unit risk 1 x 10-6 ) |
| Ethyl acetate | Mucous membrane | Irritation |
For lead, the critical organs in adults are the haemopoietic and peripheral nervous systems, where the critical effects (e.g., elevated free erythrocyte protoporphyrin concentration (FEP), increased excretion of delta-aminolevulinic acid in urine, or impaired peripheral nerve conduction) manifest when the blood lead level (an index of lead absorption in the system) approaches 200 to 300μg/l. In small children the critical organ is the central nervous system (CNS), and the symptoms of dysfunction detected with the use of a psychological test battery have been found to appear in the examined populations even at concentrations in the range of about 100μg/l Pb in blood.
A number of other definitions have been formulated which may better reflect the meaning of the notion. According to WHO (1989), the critical effect has been defined as “the first adverse effect which appears when the threshold (critical) concentration or dose is reached in the critical organ. Adverse effects, such as cancer, with no defined threshold concentration are often regarded as critical. Decision on whether an effect is critical is a matter of expert judgement.” In the International Programme on Chemical Safety (IPCS) guidelines for developing Environmental Health Criteria Documents, the critical effect is described as “the adverse effect judged to be most appropriate for determining the tolerable intake”. The latter definition has been formulated directly for the purpose of evaluating the health-based exposure limits in the general environment. In this context the most essential seems to be determining which effect can be regarded as an adverse effect. Following current terminology, the adverse effect is the “change in morphology, physiology, growth, development or lifespan of an organism which results in impairment of the capacity to compensate for additional stress or increase in susceptibility to the harmful effects of other environmental influences. Decision on whether or not any effect is adverse requires expert judgement.”
Figure 1 displays hypothetical dose-response curves for different effects. In the case of exposure to lead, A can represent a subcritical effect (inhibition of erythrocyte ALA-dehydratase), B the critical effect (an increase in erythrocyte zinc protoporphyrin or increase in the excretion of delta-aminolevulinic acid, C the clinical effect (anaemia) and D the fatal effect (death). For lead exposure there is abundant evidence illustrating how particular effects of exposure are dependent on lead concentration in blood (practical counterpart of the dose), either in the form of the dose-response relationship or in relation to different variables (sex, age, etc.). Determining the critical effects and the dose-response relationship for such effects in humans makes it possible to predict the frequency of a given effect for a given dose or its counterpart (concentration in biological material) in a certain population.
Figure 1. Hypothetical dose-response curves for various effects
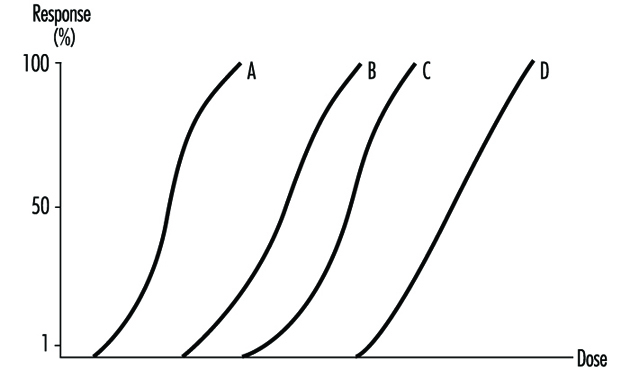
The critical effects can be of two types: those considered to have a threshold and those for which there may be some risk at any exposure level (non-threshold, genotoxic carcinogens and germ mutagens). Whenever possible, appropriate human data should be used as a basis for the risk assessment. In order to determine the threshold effects for the general population, assumptions concerning the exposure level (tolerable intake, biomarkers of exposure) have to be made such that the frequency of the critical effect in the population exposed to a given hazardous agent corresponds to the frequency of that effect in the general population. In lead exposure, the maximum recommended blood lead concentration for the general population (200μg/l, median below 100μg/l) (WHO 1987) is practically below the threshold value for the assumed critical effect—the elevated free erythrocyte protoporphyrin level, although it is not below the level associated with effects on the CNS in children or blood pressure in adults. In general, if data from well-conducted human population studies defining a no observed adverse effect level are the basis for safety evaluation, then the uncertainty factor of ten has been considered appropriate. In the case of occupational exposure the critical effects may refer to a certain part of the population (e.g. 10%). Accordingly, in occupational lead exposure the recommended health-based concentration of blood lead has been adopted to be 400mg/l in men where a 10% response level for ALA-U of 5mg/l occurred at PbB concentrations of about 300 to 400mg/l. For the occupational exposure to cadmium (assuming the increased urinary excretion of low-weight proteins to be the critical effect), the level of 200ppm cadmium in renal cortex has been regarded as the admissible value, for this effect has been observed in 10% of the exposed population. Both these values are under consideration for lowering, in many countries, at the present time (i.e.,1996).
There is no clear consensus on appropriate methodology for the risk assessment of chemicals for which the critical effect may not have a threshold, such as genotoxic carcinogens. A number of approaches based largely on characterization of the dose- response relationship have been adopted for the assessment of such effects. Owing to the lack of socio-political acceptance of health risk caused by carcinogens in such documents as the Air Quality Guidelines for Europe (WHO 1987), only the values such as the unit lifetime risk (i.e., the risk associated with lifetime exposure to 1μg/m3 of the hazardous agent) are presented for non-threshold effects (see “Regulatory toxicology”).
Presently, the basic step in undertaking activities for risk assessment is determining the critical organ and critical effects. The definitions of both the critical and adverse effect reflect the responsibility of deciding which of the effects within a given organ or system should be regarded as critical, and this is directly related to the subsequent determination of recommended values for a given chemical in the general environment—for example, Air Quality Guidelines for Europe (WHO 1987) or health-based limits in occupational exposure (WHO 1980). Determining the critical effect from within the range of subcritical effects may lead to a situation where the recommended limits on toxic chemicals concentration in the general or occupational environment may be in practice impossible to maintain. Regarding as critical an effect that may overlap the early clinical effects may bring about the adoption of the values for which adverse effects may develop in some part of the population. The decision whether or not a given effect should be considered critical remains the responsibility of expert groups who specialize in toxicity and risk assessment.
Effect of Age, Sex and Other Factors
There are often large differences among humans in the intensity of response to toxic chemicals, and variations in susceptibility of an individual over a lifetime. These can be attributed to a variety of factors capable of influencing absorption rate, distribution in the body, biotransformation and/or excretion rate of a particular chemical. Apart from the known hereditary factors which have been clearly demonstrated to be linked with increased susceptibility to chemical toxicity in humans (see “Genetic determinants of toxic response”), other factors include: constitutional characteristics related to age and sex; pre-existing disease states or a reduction in organ function (non-hereditary, i.e., acquired); dietary habits, smoking, alcohol consumption and use of medications; concomitant exposure to biotoxins (various micro- organisms) and physical factors (radiation, humidity, extremely low or high temperatures or barometric pressures particularly relevant to the partial pressure of a gas), as well as concomitant physical exercise or psychological stress situations; previous occupational and/or environmental exposure to a particular chemical, and in particular concomitant exposure to other chemicals, not necessarily toxic (e.g., essential metals). The possible contributions of the aforementioned factors in either increasing or decreasing susceptibility to adverse health effects, as well as the mechanisms of their action, are specific for a particular chemical. Therefore only the most common factors, basic mechanisms and a few characteristic examples will be presented here, whereas specific information concerning each particular chemical can be found in elsewhere in this Encyclopaedia.
According to the stage at which these factors act (absorption, distribution, biotransformation or excretion of a particular chemical), the mechanisms can be roughly categorized according to two basic consequences of interaction: (1) a change in the quantity of the chemical in a target organ, that is, at the site(s) of its effect in the organism (toxicokinetic interactions), or (2) a change in the intensity of a specific response to the quantity of the chemical in a target organ (toxicodynamic interactions). The most common mechanisms of either type of interaction are related to competition with other chemical(s) for binding to the same compounds involved in their transport in the organism (e.g., specific serum proteins) and/or for the same biotransformation pathway (e.g., specific enzymes) resulting in a change in the speed or sequence between initial reaction and final adverse health effect. However, both toxicokinetic and toxicodynamic interactions may influence individual susceptibility to a particular chemical. The influence of several concomitant factors can result in either: (a) additive effects—the intensity of the combined effect is equal to the sum of the effects produced by each factor separately, (b) synergistic effects—the intensity of the combined effect is greater than the sum of the effects produced by each factor separately, or (c) antagonistic effects—the intensity of the combined effect is smaller than the sum of the effects produced by each factor separately.
The quantity of a particular toxic chemical or characteristic metabolite at the site(s) of its effect in the human body can be more or less assessed by biological monitoring, that is, by choosing the correct biological specimen and optimal timing of specimen sampling, taking into account biological half-lives for a particular chemical in both the critical organ and in the measured biological compartment. However, reliable information concerning other possible factors that might influence individual susceptibility in humans is generally lacking, and consequently the majority of knowledge regarding the influence of various factors is based on experimental animal data.
It should be stressed that in some cases relatively large differences exist between humans and other mammals in the intensity of response to an equivalent level and/or duration of exposure to many toxic chemicals; for example, humans appear to be considerably more sensitive to the adverse health effects of several toxic metals than are rats (commonly used in experimental animal studies). Some of these differences can be attributed to the fact that the transportation, distribution and biotransformation pathways of various chemicals are greatly dependent on subtle changes in the tissue pH and the redox equilibrium in the organism (as are the activities of various enzymes), and that the redox system of the human differs considerably from that of the rat.
This is obviously the case regarding important antioxidants such as vitamin C and glutathione (GSH), which are essential for maintaining redox equilibrium and which have a protective role against the adverse effects of the oxygen- or xenobiotic-derived free radicals which are involved in a variety of pathological conditions (Kehrer 1993). Humans cannot auto-synthesize vitamin C, contrary to the rat, and levels as well as the turnover rate of erythrocyte GSH in humans are considerably lower than that in the rat. Humans also lack some of the protective antioxidant enzymes, compared to the rat or other mammals (e.g., GSH- peroxidase is considered to be poorly active in human sperm). These examples illustrate the potentially greater vulnerability to oxidative stress in humans (particularly in sensitive cells, e.g., apparently greater vulnerability of the human sperm to toxic influences than that of the rat), which can result in different response or greater susceptibility to the influence of various factors in humans compared to other mammals (Telišman 1995).
Influence of Age
Compared to adults, very young children are often more susceptible to chemical toxicity because of their relatively greater inhalation volumes and gastrointestinal absorption rate due to greater permeability of the intestinal epithelium, and because of immature detoxification enzyme systems and a relatively smaller excretion rate of toxic chemicals. The central nervous system appears to be particularly susceptible at the early stage of development with regard to neurotoxicity of various chemicals, for example, lead and methylmercury. On the other hand, the elderly may be susceptible because of chemical exposure history and increased body stores of some xenobiotics, or pre-existing compromised function of target organs and/or relevant enzymes resulting in lowered detoxification and excretion rate. Each of these factors can contribute to weakening of the body’s defences—a decrease in reserve capacity, causing increased susceptibility to subsequent exposure to other hazards. For example, the cytochrome P450 enzymes (involved in the biotransformation pathways of almost all toxic chemicals) can be either induced or have lowered activity because of the influence of various factors over a lifetime (including dietary habits, smoking, alcohol, use of medications and exposure to environmental xenobiotics).
Influence of Sex
Gender-related differences in susceptibility have been described for a large number of toxic chemicals (approximately 200), and such differences are found in many mammalian species. It appears that males are generally more susceptible to renal toxins and females to liver toxins. The causes of the different response between males and females have been related to differences in a variety of physiological processes (e.g., females are capable of additional excretion of some toxic chemicals through menstrual blood loss, breast milk and/or transfer to the foetus, but they experience additional stress during pregnancy, delivery and lactation), enzyme activities, genetic repair mechanisms, hormonal factors, or the presence of relatively larger fat depots in females, resulting in greater accumulation of some lipophilic toxic chemicals, such as organic solvents and some medications.
Influence of Dietary Habits
Dietary habits have an important influence on susceptibility to chemical toxicity, mostly because adequate nutrition is essential for the functioning of the body’s chemical defence system in maintaining good health. Adequate intake of essential metals (including metalloids) and proteins, especially the sulphur-containing amino acids, is necessary for the biosynthesis of various detoxificating enzymes and the provision of glycine and glutathione for conjugation reactions with endogenous and exogenous compounds. Lipids, especially phospholipids, and lipotropes (methyl group donors) are necessary for the synthesis of biological membranes. Carbohydrates provide the energy required for various detoxification processes and provide glucuronic acid for conjugation of toxic chemicals and their metabolites. Selenium (an essential metalloid), glutathione, and vitamins such as vitamin C (water soluble), vitamin E and vitamin A (lipid soluble), have an important role as antioxidants (e.g., in controlling lipid peroxidation and maintaining integrity of cellular membranes) and free-radical scavengers for protection against toxic chemicals. In addition, various dietary constituents (protein and fibre content, minerals, phosphates, citric acid, etc.) as well as the amount of food consumed can greatly influence the gastrointestinal absorption rate of many toxic chemicals (e.g., the average absorption rate of soluble lead salts taken with meals is approximately eight per cent, as opposed to approximately 60% in fasting subjects). However, diet itself can be an additional source of individual exposure to various toxic chemicals (e.g., considerably increased daily intakes and accumulation of arsenic, mercury, cadmium and/or lead in subjects who consume contaminated seafood).
Influence of Smoking
The habit of smoking can influence individual susceptibility to many toxic chemicals because of the variety of possible interactions involving the great number of compounds present in cigarette smoke (especially polycyclic aromatic hydrocarbons, carbon monoxide, benzene, nicotine, acrolein, some pesticides, cadmium, and, to a lesser extent, lead and other toxic metals, etc.), some of which are capable of accumulating in the human body over a lifetime, including pre-natal life (e.g., lead and cadmium). The interactions occur mainly because various toxic chemicals compete for the same binding site(s) for transport and distribution in the organism and/or for the same biotransformation pathway involving particular enzymes. For example, several cigarette smoke constituents can induce cytochrome P450 enzymes, whereas others can depress their activity, and thus influence the common biotransformation pathways of many other toxic chemicals, such as organic solvents and some medications. Heavy cigarette smoking over a long period can considerably reduce the body’s defence mechanisms by decreasing reserve capacity to cope with the adverse influence of other life-style factors.
Influence of Alcohol
Consumption of alcohol (ethanol) can influence susceptibility to many toxic chemicals in several ways. It can influence the absorption rate and distribution of certain chemicals in the body—for example, increase the gastrointestinal absorption rate of lead, or decrease the pulmonary absorption rate of mercury vapour by inhibiting oxidation which is necessary for retention of inhaled mercury vapour. Ethanol can also influence susceptibility to various chemicals through short-term changes in tissue pH and increase in the redox potential resulting from ethanol metabolism, as both ethanol oxidizing to acetaldehyde and acetaldehyde oxidizing to acetate produce an equivalent of reduced nicotinamide adenine dinucleotide (NADH) and hydrogen (H+). Because the affinity of both essential and toxic metals and metalloids for binding to various compounds and tissues is influenced by pH and changes in the redox potential (Telišman 1995), even a moderate intake of ethanol may result in a series of consequences such as: (1) redistribution of long-term accumulated lead in the human organism in favour of a biologically active lead fraction, (2) replacement of essential zinc by lead in zinc-containing enzyme(s), thus affecting enzyme activity, or influence of mobil- ized lead on the distribution of other essential metals and metalloids in the organism such as calcium, iron, copper and selenium, (3) increased urinary excretion of zinc and so on. The effect of possible aforementioned events can be augmented due to the fact that alcoholic beverages can contain an appreciable amount of lead from vessels or processing (Prpic-Majic et al. 1984; Telišman et al. 1984; 1993).
Another common reason for ethanol-related changes in susceptibility is that many toxic chemicals, for example, various organic solvents, share the same biotransformation pathway involving the cytochrome P450 enzymes. Depending on the intensity of exposure to organic solvents as well as the quantity and frequency of ethanol ingestion (i.e., acute or chronic alcohol consumption), ethanol can either decrease or increase biotransformation rates of various organic solvents and thus influence their toxicity (Sato 1991).
Influence of Medications
The common use of various medications can influence susceptibility to toxic chemicals mainly because many drugs bind to serum proteins and thus influence the transport, distribution or excretion rate of various toxic chemicals, or because many drugs are capable of inducing relevant detoxifying enzymes or depressing their activity (e.g., the cytochrome P450 enzymes), thus affecting the toxicity of chemicals with the same biotransformation pathway. Characteristic for either of the mechanisms is increased urinary excretion of trichloroacetic acid (the metabolite of several chlorinated hydrocarbons) when using salicylate, sulphonamide or phenylbutazone, and an increased hepato-nephrotoxicity of carbon tetrachloride when using phenobarbital. In addition, some medications contain a considerable amount of a potentially toxic chemical, for example, the aluminium-containing antacids or preparations used for therapeutic management of the hyperphosphataemia arising in chronic renal failure.
Influence of Concomitant Exposure to Other Chemicals
The changes in susceptibility to adverse health effects due to interaction of various chemicals (i.e., possible additive, synergistic or antagonistic effects) have been studied almost exclusively in experimental animals, mostly in the rat. Relevant epidemiological and clinical studies are lacking. This is of concern particularly considering the relatively greater intensity of response or the variety of adverse health effects of several toxic chemicals in humans compared to the rat and other mammals. Apart from published data in the field of pharmacology, most data are related only to combinations of two different chemicals within specific groups, such as various pesticides, organic solvents, or essential and/or toxic metals and metalloids.
Combined exposure to various organic solvents can result in various additive, synergistic or antagonistic effects (depending on the combination of certain organic solvents, their intensity and duration of exposure), mainly due to the capability of influencing each other’s biotransformation (Sato 1991).
Another characteristic example are the interactions of both essential and/or toxic metals and metalloids, as these are involved in the possible influence of age (e.g., a lifetime body accumulation of environmental lead and cadmium), sex (e.g., common iron deficiency in women), dietary habits (e.g., increased dietary intake of toxic metals and metalloids and/or deficient dietary intake of essential metals and metalloids), smoking habit and alcohol consumption (e.g., additional exposure to cadmium, lead and other toxic metals), and use of medications (e.g., a single dose of antacid can result in a 50-fold increase in the average daily intake of aluminium through food). The possibility of various additive, synergistic or antagonistic effects of exposure to various metals and metalloids in humans can be illustrated by basic examples related to the main toxic elements (see table 1), apart from which further interactions may occur because essential elements can also influence one another (e.g., the well-known antagonistic effect of copper on the gastrointestinal absorption rate as well as the metabolism of zinc, and vice versa). The main cause of all these interactions is the competition of various metals and metalloids for the same binding site (especially the sulphhydryl group, -SH) in various enzymes, metalloproteins (especially metallothionein) and tissues (e.g., cell membranes and organ barriers). These interactions may have a relevant role in the development of several chronic diseases which are mediated through the action of free radicals and oxidative stress (Telišman 1995).
Table 1. Basic effects of possible multiple interactions concerning the main toxic and/or essential metals and matalloids in mammals
| Toxic metal or metalloid | Basic effects of the interaction with other metal or metalloid |
| Aluminium (Al) | Decreases the absorption rate of Ca and impairs the metabolism of Ca; deficient dietary Ca increases the absorption rate of Al. Impairs phosphate metabolism. Data on interactions with Fe, Zn and Cu are equivocal (i.e., the possible role of another metal as a mediator). |
| Arsenic (As) | Affects the distribution of Cu (an increase of Cu in the kidney, and a decrease of Cu in the liver, serum and urine). Impairs the metabolism of Fe (an increase of Fe in the liver with concomitant decrease in haematocrit). Zn decreases the absorption rate of inorganic As and decreases the toxicity of As. Se decreases the toxicity of As and vice versa. |
| Cadmium (Cd) | Decreases the absorption rate of Ca and impairs the metabolism of Ca; deficient dietary Ca increases the absorption rate of Cd. Impairs the phosphate metabolism, i.e., increases urinary excretion of phosphates. Impairs the metabolism of Fe; deficient dietary Fe increases the absorption rate of Cd. Affects the distribution of Zn; Zn decreases the toxicity of Cd, whereas its influence on the absorption rate of Cd is equivocal. Se decreases the toxicity of Cd. Mn decreases the toxicity of Cd at low-level exposure to Cd. Data on the interaction with Cu are equivocal (i.e., the possible role of Zn, or another metal, as a mediator). High dietary levels of Pb, Ni, Sr, Mg or Cr(III) can decrease the absorption rate of Cd. |
| Mercury (Hg) | Affects the distribution of Cu (an increase of Cu in the liver). Zn decreases the absorption rate of inorganic Hg and decreases the toxicity of Hg. Se decreases the toxicity of Hg. Cd increases the concentration of Hg in the kidney, but at the same time decreases the toxicity of Hg in the kidney (the influence ofthe Cd-induced metallothionein synthesis). |
| Lead (Pb) | Impairs the metabolism of Ca; deficient dietary Ca increases the absorption rate of inorganic Pb and increases the toxicity of Pb. Impairs the metabolism of Fe; deficient dietary Fe increases the toxicity of Pb, whereas its influence on the absorption rate of Pb is equivocal. Impairs the metabolism of Zn and increases urinary excretion of Zn; deficient dietary Zn increases the absorption rate of inorganic Pb andincreases the toxicity of Pb. Se decreases the toxicity of Pb. Data on interactions with Cu and Mg are equivocal (i.e., the possible role of Zn, or another metal, as a mediator). |
Note: Data are mostly related to experimental studies in the rat, whereas relevant clinical and epidemiological data (particularly regarding quantitative dose-response relationships) are generally lacking (Elsenhans et al. 1991; Fergusson 1990; Telišman et al. 1993).
Genetic Determinants of Toxic Response
It has long been recognized that each person’s response to environmental chemicals is different. The recent explosion in molecular biology and genetics has brought a clearer understanding about the molecular basis of such variability. Major determinants of individual response to chemicals include important differences among more than a dozen superfamilies of enzymes, collectively termed xenobiotic- (foreign to the body) or drug-metabolizing enzymes. Although the role of these enzymes has classically been regarded as detoxification, these same enzymes also convert a number of inert compounds to highly toxic intermediates. Recently, many subtle as well as gross differences in the genes encoding these enzymes have been identified, which have been shown to result in marked variations in enzyme activity. It is now clear that each individual possesses a distinct complement of xenobiotic-metabolizing enzyme activities; this diversity might be thought of as a “metabolic fingerprint”. It is the complex interplay of these many different enzyme superfamilies which ultimately determines not only the fate and the potential for toxicity of a chemical in any given individual, but also assessment of exposure. In this article we have chosen to use the cytochrome P450 enzyme superfamily to illustrate the remarkable progress made in understanding individual response to chemicals. The development of relatively simple DNA-based tests designed to identify specific gene alterations in these enzymes, is now providing more accurate predictions of individual response to chemical exposure. We hope the result will be preventive toxicology. In other words, each individual might learn about those chemicals to which he or she is particularly sensitive, thereby avoiding previously unpredictable toxicity or cancer.
Although it is not generally appreciated, human beings are exposed daily to a barrage of innumerable diverse chemicals. Many of these chemicals are highly toxic, and they are derived from a wide variety of environmental and dietary sources. The relationship between such exposures and human health has been, and continues to be, a major focus of biomedical research efforts worldwide.
What are some examples of this chemical bombardment? More than 400 chemicals from red wine have been isolated and characterized. At least 1,000 chemicals are estimated to be produced by a lighted cigarette. There are countless chemicals in cosmetics and perfumed soaps. Another major source of chemical exposure is agriculture: in the United States alone, farmlands receive more than 75,000 chemicals each year in the form of pesticides, herbicides and fertilizing agents; after uptake by plants and grazing animals, as well as fish in nearby waterways, humans (at the end of the food chain) ingest these chemicals. Two other sources of large concentrations of chemicals taken into the body include (a) drugs taken chronically and (b) exposure to hazardous substances in the workplace over a lifetime of employment.
It is now well established that chemical exposure may adversely affect many aspects of human health, causing chronic diseases and the development of many cancers. In the last decade or so, the molecular basis of many of these relationships has begun to be unravelled. In addition, the realization has emerged that humans differ markedly in their susceptibility to the harmful effects of chemical exposure.
Current efforts to predict human response to chemical exposure combine two fundamental approaches (figure 1): monitoring the extent of human exposure through biological markers (biomarkers), and predicting the likely response of an individual to a given level of exposure. Although both of these approaches are extremely important, it should be emphasized that the two are distinctly different from one another. This article will focus on the genetic factors underlying individual susceptibility to any particular chemical exposure. This field of research is broadly termed ecogenetics, or pharmacogenetics (see Kalow 1962 and 1992). Many of the recent advances in determining individual susceptibility to chemical toxicity have evolved from a greater appreciation of the processes by which humans and other mammals detoxify chemicals, and the remarkable complexity of the enzyme systems involved.
Figure 1. The interrelationships among exposure assessment, ethnic differences, age, diet, nutrition and genetic susceptibility assessment - all of which play a role in the individual risk of toxicity and cancer
We will first describe the variability of toxic responses in humans. We will then introduce some of the enzymes responsible for such variation in response, due to differences in the metabolism of foreign chemicals. Next, the history and nomenclature of the cytochrome P450 superfamily will be detailed. Five human P450 polymorphisms as well as several non-P450 polymorphisms will be briefly described; these are responsible for human differences in toxic response. We will then discuss an example to emphasize the point that genetic differences in individuals can influence exposure assessment, as determined by environmental monitoring. Lastly, we will discuss the role of these xenobiotic-metabolizing enzymes in critical life functions.
Variation in Toxic Response Among the Human Population
Toxicologists and pharmacologists commonly speak about the average lethal dose for 50% of the population (LD50), the average maximal tolerated dose for 50% of the population (MTD50), and the average effective dose of a particular drug for 50% of the population (ED50). However, how do these doses affect each of us on an individual basis? In other words, a highly sensitive individual may be 500 times more affected or 500 times more likely to be affected than the most resistant individual in a population; for these people, the LD50 (and MTD50 and ED50) values would have little meaning. LD50, MTD50 and ED50 values are only relevant when referring to the population as a whole.
Figure 2 illustrates a hypothetical dose-response relationship for a toxic response by individuals in any given population. This generic diagram might represent bronchogenic carcinoma in response to the number of cigarettes smoked, chloracne as a function of dioxin levels in the workplace, asthma as a function of air concentrations of ozone or aldehyde, sunburn in response to ultraviolet light, decreased clotting time as a function of aspirin intake, or gastrointestinal distress in response to the number of jalapeño peppers consumed. Generally, in each of these instances, the greater the exposure, the greater the toxic response. Most of the population will exhibit the mean and standard deviation of toxic response as a function of dose. The “resistant outlier” (lower right in figure 2) is an individual having less of a response at higher doses or exposures. A “sensitive outlier” (upper left) is an individual having an exaggerated response to a relatively small dose or exposure. These outliers, with extreme differences in response compared to the majority of individuals in the population, may represent important genetic variants that can help scientists in attempting to understand the underlying molecular mechanisms of a toxic response.
Figure 2. Generic relationship between any toxic response and the dose of any environmental, chemical or physical agent
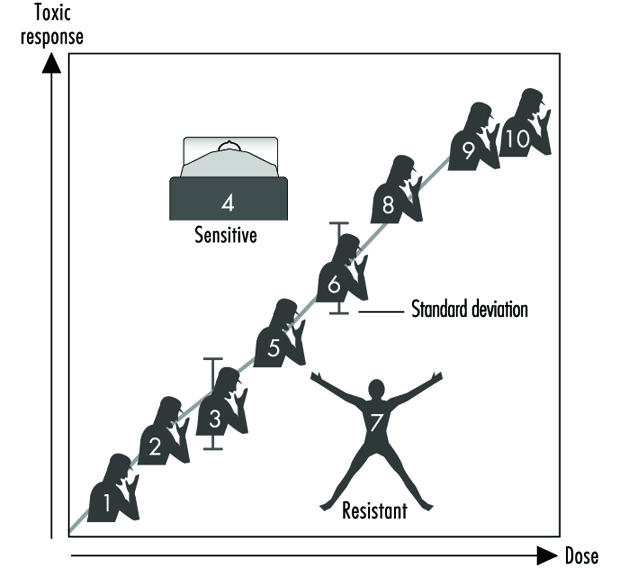
Using these outliers in family studies, scientists in a number of laboratories have begun to appreciate the importance of Mendelian inheritance for a given toxic response. Subsequently, one can then turn to molecular biology and genetic studies to pinpoint the underlying mechanism at the gene level (genotype) responsible for the environmentally caused disease (phenotype).
Xenobiotic- or Drug-metabolizing Enzymes
How does the body respond to the myriad of exogenous chemicals to which we are exposed? Humans and other mammals have evolved highly complex metabolic enzyme systems comprising more than a dozen distinct superfamilies of enzymes. Almost every chemical to which humans are exposed will be modified by these enzymes, in order to facilitate removal of the foreign substance from the body. Collectively, these enzymes are frequently referred to as drug-metabolizing enzymes or xenobiotic-metabolizing enzymes. Actually, both terms are misnomers. First, many of these enzymes not only metabolize drugs but hundreds of thousands of environmental and dietary chemicals. Second, all of these enzymes also have normal body compounds as substrates; none of these enzymes metabolizes only foreign chemicals.
For more than four decades, the metabolic processes mediated by these enzymes have commonly been classified as either Phase I or Phase II reactions (figure 3). Phase I (“functionalization”) reactions generally involve relatively minor structural modifications of the parent chemical via oxidation, reduction or hydrolysis in order to produce a more water-soluble metabolite. Frequently, Phase I reactions provide a “handle” for further modification of a compound by subsequent Phase II reactions. Phase I reactions are primarily mediated by a superfamily of highly versatile enzymes, collectively termed cytochromes P450, although other enzyme superfamilies can also be involved (figure 4).
Figure 3. The classical designation of Phase I and Phase II xenobiotic- or drug-metabolizing enzymes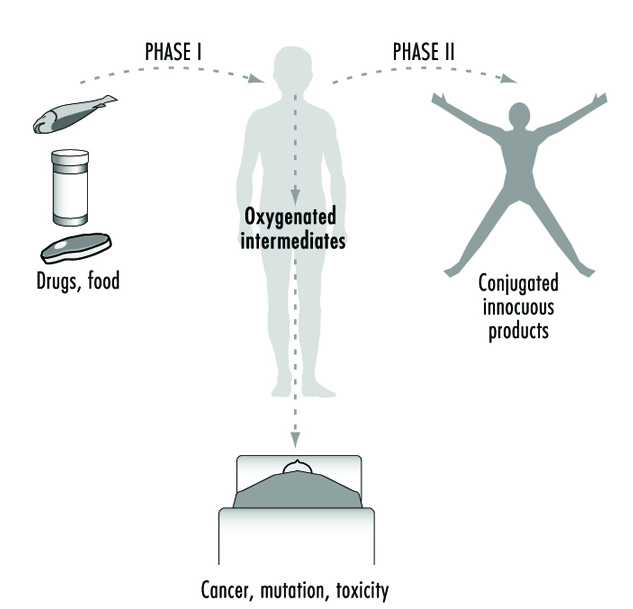
Figure 4. Examples of drug-metabolizing enzymes
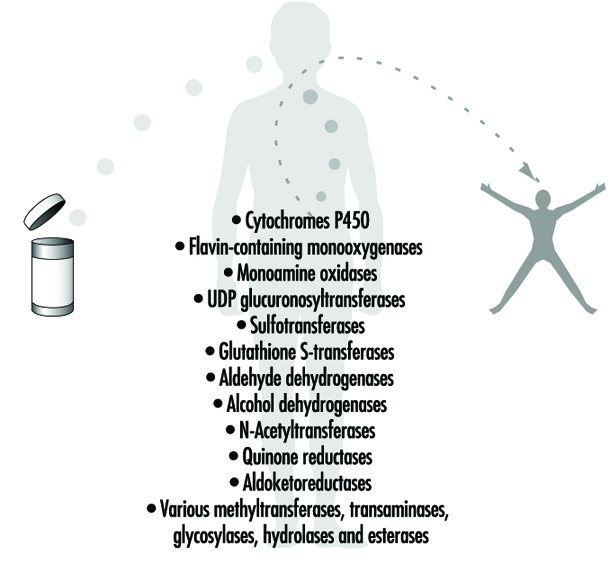
Phase II reactions involve the coupling of a water-soluble endogenous molecule to a chemical (parent chemical or Phase I metabolite) in order to facilitate excretion. Phase II reactions are frequently termed “conjugation” or “derivatization” reactions. The enzyme superfamilies catalyzing Phase II reactions are generally named according to the endogenous conjugating moiety involved: for example, acetylation by the N-acetyltransferases, sulphation by the sulphotransferases, glutathione conjugation by the glutathione transferases, and glucuronidation by the UDP glucuronosyltransferases (figure 4). Although the major organ of drug metabolism is the liver, the levels of some drug- metabolizing enzymes are quite high in the gastrointestinal tract, gonads, lung, brain and kidney, and such enzymes are undoubtedly present to some extent in every living cell.
Xenobiotic-metabolizing Enzymes Represent Double-edged Swords
As we learn more about the biological and chemical processes leading to human health aberrations, it has become increasingly evident that drug-metabolizing enzymes function in an ambivalent manner (figure 3). In the majority of cases, lipid-soluble chemicals are converted to more readily excreted water-soluble metabolites. However, it is clear that on many occasions the same enzymes are capable of transforming other inert chemicals into highly reactive molecules. These intermediates can then interact with cellular macromolecules such as proteins and DNA. Thus, for each chemical to which humans are exposed, there exists the potential for the competing pathways of metabolic activation and detoxification.
Brief Review of Genetics
In human genetics, each gene (locus) is located on one of the 23 pairs of chromosomes. The two alleles (one present on each chromosome of the pair) can be the same, or they can be different from one another. For example, the B and b alleles, in which B (brown eyes) is dominant over b (blue eyes): individuals of the brown-eyed phenotype can have either the BB or Bb genotypes, whereas individuals of the blue-eyed phenotype can only have the bb genotype.
A polymorphism is defined as two or more stably inherited phenotypes (traits)—derived from the same gene(s)—that are maintained in the population, often for reasons not necessarily obvious. For a gene to be polymorphic, the gene product must not be essential for development, reproductive vigour or other critical life processes. In fact, a “balanced polymorphism,” wherein the heterozygote has a distinct survival advantage over either homozygote (e.g., resistance to malaria, and the sickle-cell haemoglobin allele) is a common explanation for maintaining an allele in the population at otherwise unexplained high frequencies (see Gonzalez and Nebert 1990).
Human Polymorphisms of Xenobiotic-metabolizing Enzymes
Genetic differences in the metabolism of various drugs and environmental chemicals have been known for more than four decades (Kalow 1962 and 1992). These differences are frequently referred to as pharmacogenetic or, more broadly, ecogenetic polymorphisms. These polymorphisms represent variant alleles that occur at a relatively high frequency in the population and are generally associated with aberrations in enzyme expression or function. Historically, polymorphisms were usually identified following unexpected responses to therapeutic agents. More recently, recombinant DNA technology has enabled scientists to identify the precise alterations in genes that are responsible for some of these polymorphisms. Polymorphisms have now been characterized in many drug-metabolizing enzymes—including both Phase I and Phase II enzymes. As more and more polymorphisms are identified, it is becoming increasingly apparent that each individual may possess a distinct complement of drug-metabolizing enzymes. This diversity might be described as a “metabolic fingerprint”. It is the complex interplay of the various drug- metabolizing enzyme superfamilies within any individual that will ultimately determine his or her particular response to a given chemical (Kalow 1962 and 1992; Nebert 1988; Gonzalez and Nebert 1990; Nebert and Weber 1990).
Expressing Human Xenobiotic-metabolizingEnzymes in Cell Culture
How might we develop better predictors of human toxic responses to chemicals? Advances in defining the multiplicity of drug-metabolizing enzymes must be accompanied by precise knowledge as to which enzymes determine the metabolic fate of individual chemicals. Data gleaned from laboratory rodent studies have certainly provided useful information. However, significant interspecies differences in xenobiotic-metabolizing enzymes necessitate caution in extrapolating data to human populations. To overcome this difficulty, many laboratories have developed systems in which various cell lines in culture can be engineered to produce functional human enzymes that are stable and in high concentrations (Gonzalez, Crespi and Gelboin 1991). Successful production of human enzymes has been achieved in a variety of diverse cell lines from sources including bacteria, yeast, insects and mammals.
In order to define the metabolism of chemicals even more accurately, multiple enzymes have also been successfully produced in a single cell line (Gonzalez, Crespi and Gelboin 1991). Such cell lines provide valuable insights into the precise enzymes involved in the metabolic processing of any given compound and likely toxic metabolites. If this information can then be combined with knowledge regarding the presence and level of an enzyme in human tissues, these data should provide valuable predictors of response.
Cytochrome P450
History and nomenclature
The cytochrome P450 superfamily is one of the most studied drug-metabolizing enzyme superfamilies, having a great deal of individual variability in response to chemicals. Cytochrome P450 is a convenient generic term used to describe a large superfamily of enzymes pivotal in the metabolism of innumerable endogenous and exogenous substrates. The term cytochrome P450 was first coined in 1962 to describe an unknown pigment in cells which, when reduced and bound with carbon monoxide, produced a characteristic absorption peak at 450 nm. Since the early 1980s, cDNA cloning technology has resulted in remarkable insights into the multiplicity of cytochrome P450 enzymes. To date, more than 400 distinct cytochrome P450 genes have been identified in animals, plants, bacteria and yeast. It has been estimated that any one mammalian species, such as humans, may possess 60 or more distinct P450 genes (Nebert and Nelson 1991). The multiplicity of P450 genes has necessitated the development of a standardized nomenclature system (Nebert et al. 1987; Nelson et al. 1993). First proposed in 1987 and updated on a biannual basis, the nomenclature system is based on divergent evolution of amino acid sequence comparisons between P450 proteins. The P450 genes are divided into families and subfamilies: enzymes within a family display greater than 40% amino acid similarity, and those within the same subfamily display 55% similarity. P450 genes are named with the root symbol CYP followed by an arabic numeral designating the P450 family, a letter denoting the subfamily, and a further arabic numeral designating the individual gene (Nelson et al. 1993; Nebert et al. 1991). Thus, CYP1A1 represents P450 gene 1 in family 1 and subfamily A.
As of February 1995, there are 403 CYP genes in the database, composed of 59 families and 105 sub- families. These include eight lower eukaryotic families, 15 plant families, and 19 bacterial families. The 15 human P450 gene families comprise 26 subfamilies, 22 of which have been mapped to chromosomal locations throughout most of the genome. Some sequences are clearly orthologous across many species—for example, only one CYP17 (steroid 17α-hydroxylase) gene has been found in all vertebrates examined to date; other sequences within a subfamily are highly duplicated, making the identification of orthologous pairs impossible (e.g., the CYP2C subfamily). Interestingly, human and yeast share an orthologous gene in the CYP51 family. Numerous comprehensive reviews are available for readers seeking further information on the P450 superfamily (Nelson et al. 1993; Nebert et al. 1991; Nebert and McKinnon 1994; Guengerich 1993; Gonzalez 1992).
The success of the P450 nomenclature system has resulted in similar terminology systems being developed for the UDP glucuronosyltransferases (Burchell et al. 1991) and flavin-containing mono-oxygenases (Lawton et al. 1994). Similar nomenclature systems based on divergent evolution are also under development for several other drug-metabolizing enzyme superfamilies (e.g., sulphotransferases, epoxide hydrolases and aldehyde dehydrogenases).
Recently, we divided the mammalian P450 gene superfamily into three groups (Nebert and McKinnon 1994)—those involved principally with foreign chemical metabolism, those involved in the synthesis of various steroid hormones, and those participating in other important endogenous functions. It is the xenobiotic-metabolizing P450 enzymes that assume the most significance for prediction of toxicity.
Xenobiotic-metabolizing P450 enzymes
P450 enzymes involved in the metabolism of foreign compounds and drugs are almost always found within families CYP1, CYP2, CYP3 and CYP4. These P450 enzymes catalyze a wide variety of metabolic reactions, with a single P450 often capable of meta-bolizing many different compounds. In addition, multiple P450 enzymes may metabolize a single compound at different sites. Also, a compound may be metabolized at the same, single site by several P450s, although at varying rates.
A most important property of the drug-metabolizing P450 enzymes is that many of these genes are inducible by the very substances which serve as their substrates. On the other hand, other P450 genes are induced by nonsubstrates. This phenomenon of enzyme induction underlies many drug-drug interactions of therapeutic importance.
Although present in many tissues, these particular P450 enzymes are found in relatively high levels in the liver, the primary site of drug metabolism. Some of the xenobiotic-metabolizing P450 enzymes exhibit activity toward certain endogenous substrates (e.g., arachidonic acid). However, it is generally believed that most of these xenobiotic-metabolizing P450 enzymes do not play important physiological roles—although this has not been established experimentally as yet. The selective homozygous disruption, or “knock-out,” of individual xenobiotic-metabolizing P450 genes by means of gene targeting methodologies in mice is likely to provide unequivocal information soon with regard to physiological roles of the xenobiotic-metabolizing P450s (for a review of gene targeting, see Capecchi 1994).
In contrast to P450 families encoding enzymes involved primarily in physiological processes, families encoding xenobiotic-metabolizing P450 enzymes display marked species specificity and frequently contain many active genes per subfamily (Nelson et al. 1993; Nebert et al. 1991). Given the apparent lack of physiological substrates, it is possible that P450 enzymes in families CYP1, CYP2, CYP3 and CYP4 that have appeared in the past several hundred million years have evolved as a means of detoxifying foreign chemicals encountered in the environment and diet. Clearly, evolution of the xenobiotic-metabolizing P450s would have occurred over a time period which far precedes the synthesis of most of the synthetic chemicals to which humans are now exposed. The genes in these four gene families may have evolved and diverged in animals due to their exposure to plant metabolites during the last 1.2 billion years—a process descriptively termed “animal-plant warfare” (Gonzalez and Nebert 1990). Animal-plant warfare is the phenomenon in which plants developed new chemicals (phytoalexins) as a defence mechanism in order to prevent ingestion by animals, and animals, in turn, responded by developing new P450 genes to accommodate the diversifying substrates. Providing further impetus to this proposal are the recently described examples of plant-insect and plant-fungus chemical warfare involving P450 detoxification of toxic substrates (Nebert 1994).
The following is a brief introduction to several of the human xenobiotic-metabolizing P450 enzyme polymorphisms in which genetic determinants of toxic response are believed to be of high significance. Until recently, P450 polymorphisms were generally suggested by unexpected variance in patient response to administered therapeutic agents. Several P450 polymorphisms are indeed named according to the drug with which the polymorphism was first identified. More recently, research efforts have focused on identification of the precise P450 enzymes involved in the metabolism of chemicals for which variance is observed and the precise characterization of the P450 genes involved. As described earlier, the measurable activity of a P450 enzyme towards a model chemical can be called the phenotype. Allelic differences in a P450 gene for each individual is termed the P450 genotype. As more and more scrutiny is applied to the analysis of P450 genes, the precise molecular basis of previously documented phenotypic variance is becoming clearer.
The CYP1A subfamily
The CYP1A subfamily comprises two enzymes in humans and all other mammals: these are designated CYP1A1 and CYP1A2 under standard P450 nomenclature. These enzymes are of considerable interest, because they are involved in the metabolic activation of many procarcinogens and are also induced by several compounds of toxicological concern, including dioxin. For example, CYP1A1 metabolically activates many compounds found in cigarette smoke. CYP1A2 metabolically activates many arylamines—associated with urinary bladder cancer—found in the chemical dye industry. CYP1A2 also metabolically activates 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a tobacco-derived nitrosamine. CYP1A1 and CYP1A2 are also found at higher levels in the lungs of cigarette smokers, due to induction by polycyclic hydrocarbons present in the smoke. The levels of CYP1A1 and CYP1A2 activity are therefore considered to be important determinants of individual response to many potentially toxic chemicals.
Toxicological interest in the CYP1A subfamily was greatly intensified by a 1973 report correlating the level of CYP1A1 inducibility in cigarette smokers with individual susceptibility to lung cancer (Kellermann, Shaw and Luyten-Kellermann 1973). The molecular basis of CYP1A1 and CYP1A2 induction has been a major focus of numerous laboratories. The induction process is mediated by a protein termed the Ah receptor to which dioxins and structurally related chemicals bind. The name Ah is derived from the aryl hydrocarbon nature of many CYP1A inducers. Interestingly, differences in the gene encoding the Ah receptor between strains of mice result in marked differences in chemical response and toxicity. A polymorphism in the Ah receptor gene also appears to occur in humans: approximately one-tenth of the population displays high induction of CYP1A1 and may be at greater risk than the other nine-tenths of the population for development of certain chemically induced cancers. The role of the Ah receptor in the control of enzymes in the CYP1A subfamily, and its role as a determinant of human response to chemical exposure, has been the subject of several recent reviews (Nebert, Petersen and Puga 1991; Nebert, Puga and Vasiliou 1993).
Are there other polymorphisms that might control the level of CYP1A proteins in a cell? A polymorphism in the CYP1A1 gene has also been identified, and this appears to influence lung cancer risk amongst Japanese cigarette smokers, although this same polymorphism does not appear to influence risk in other ethnic groups (Nebert and McKinnon 1994).
CYP2C19
Variations in the rate at which individuals metabolize the anticonvulsant drug (S)-mephenytoin have been well documented for many years (Guengerich 1989). Between 2% and 5% of Caucasians and as many as 25% of Asians are deficient in this activity and may be at greater risk of toxicity from the drug. This enzyme defect has long been known to involve a member of the human CYP2C subfamily, but the precise molecular basis of this deficiency has been the subject of considerable controversy. The major reason for this difficulty was the six or more genes in the human CYP2C subfamily. It was recently demonstrated, however, that a single-base mutation in the CYP2C19 gene is the primary cause of this deficiency (Goldstein and de Morais 1994). A simple DNA test, based on the polymerase chain reaction (PCR), has also been developed to identify this mutation rapidly in human populations (Goldstein and de Morais 1994).
CYP2D6
Perhaps the most extensively characterized variation in a P450 gene is that involving the CYP2D6 gene. More than a dozen examples of mutations, rearrangements and deletions affecting this gene have been described (Meyer 1994). This polymorphism was first suggested 20 years ago by clinical variability in patients’ response to the antihypertensive agent debrisoquine. Alterations in the CYP2D6 gene giving rise to altered enzyme activity are therefore collectively termed the debrisoquine polymorphism.
Prior to the advent of DNA-based studies, individuals had been classified as poor or extensive metabolizers (PMs, EMs) of debrisoquine based on metabolite concentrations in urine samples. It is now clear that alterations in the CYP2D6 gene may result in individuals displaying not only poor or extensive debrisoquine metabolism, but also ultrarapid metabolism. Most alterations in the CYP2D6 gene are associated with partial or total deficiency of enzyme function; however, individuals in two families have recently been described who possess multiple functional copies of the CYP2D6 gene, giving rise to ultrarapid metabolism of CYP2D6 substrates (Meyer 1994). This remarkable observation provides new insights into the wide spectrum of CYP2D6 activity previously observed in population studies. Alterations in CYP2D6 function are of particular significance, given the more than 30 commonly prescribed drugs metabolized by this enzyme. An individual’s CYP2D6 function is therefore a major determinant of both therapeutic and toxic response to administered therapy. Indeed, it has recently been argued that consideration of a patient’s CYP2D6 status is necessary for the safe use of both psychiatric and cardiovascular drugs.
The role of the CYP2D6 polymorphism as a determinant of individual susceptibility to human diseases such as lung cancer and Parkinson’s disease has also been the subject of intense study (Nebert and McKinnon 1994; Meyer 1994). While conclusions are difficult to define given the diverse nature of the study protocols utilized, the majority of studies appear to indicate an association between extensive metabolizers of debrisoquine (EM phenotype) and lung cancer. The reasons for such an association are presently unclear. However, the CYP2D6 enzyme has been shown to metabolize NNK, a tobacco-derived nitrosamine.
As DNA-based assays improve—enabling even more accurate assessment of CYP2D6 status—it is anticipated that the precise relationship of CYP2D6 to disease risk will be clarified. Whereas the extensive metabolizer may be linked with susceptibility to lung cancer, the poor metabolizer (PM phenotype) appears to be associated with Parkinson’s disease of unknown cause. Whereas these studies are also difficult to compare, it appears that PM individuals having a diminished capacity to metabolize CYP2D6 substrates (e.g., debrisoquine) have a 2- to 2.5-fold increase in risk of developing Parkinson’s disease.
CYP2E1
The CYP2E1 gene encodes an enzyme that metabolizes many chemicals, including drugs and many low-molecular-weight carcinogens. This enzyme is also of interest because it is highly inducible by alcohol and may play a role in liver injury induced by chemicals such as chloroform, vinyl chloride and carbon tetrachloride. The enzyme is primarily found in the liver, and the level of enzyme varies markedly between individuals. Close scrutiny of the CYP2E1 gene has resulted in the identification of several polymorphisms (Nebert and McKinnon 1994). A relationship has been reported between the presence of certain structural variations in the CYP2E1 gene and apparent lowered lung cancer risk in some studies; however, there are clear interethnic differences which require clarification of this possible relationship.
The CYP3A subfamily
In humans, four enzymes have been identified as members of the CYP3A subfamily due to their similarity in amino acid sequence. The CYP3A enzymes metabolize many commonly prescribed drugs such as erythromycin and cyclosporin. The carcinogenic food contaminant aflatoxin B1 is also a CYP3A substrate. One member of the human CYP3A subfamily, designated CYP3A4, is the principal P450 in human liver as well as being present in the gastrointestinal tract. As is true for many other P450 enzymes, the level of CYP3A4 is highly variable between individuals. A second enzyme, designated CYP3A5, is found in only approximately 25% of livers; the genetic basis of this finding has not been elucidated. The importance of CYP3A4 or CYP3A5 variability as a factor in genetic determinants of toxic response has not yet been established (Nebert and McKinnon 1994).
Non-P450 Polymorphisms
Numerous polymorphisms also exist within other xenobiotic-metabolizing enzyme superfamilies (e.g., glutathione transferases, UDP glucuronosyltransferases, para-oxonases, dehydrogenases, N-acetyltransferases and flavin-containing mono-oxygenases). Because the ultimate toxicity of any P450-generated intermediate is dependent on the efficiency of subsequent Phase II detoxification reactions, the combined role of multiple enzyme polymorphisms is important in determining susceptibility to chemically induced diseases. The metabolic balance between Phase I and Phase II reactions (figure 3) is therefore likely to be a major factor in chemically induced human diseases and genetic determinants of toxic response.
The GSTM1 gene polymorphism
A well studied example of a polymorphism in a Phase II enzyme is that involving a member of the glutathione S-transferase enzyme superfamily, designated GST mu or GSTM1. This particular enzyme is of considerable toxicological interest because it appears to be involved in the subsequent detoxification of toxic metabolites produced from chemicals in cigarette smoke by the CYP1A1 enzyme. The identified polymorphism in this glutathione transferase gene involves a total absence of functional enzyme in as many as half of all Caucasians studied. This lack of a Phase II enzyme appears to be associated with increased susceptibility to lung cancer. By grouping individuals on the basis of both variant CYP1A1 genes and the deletion or presence of a functional GSTM1 gene, it has been demonstrated that the risk of developing smoking-induced lung cancer varies significantly (Kawajiri, Watanabe and Hayashi 1994). In particular, individuals displaying one rare CYP1A1 gene alteration, in combination with an absence of the GSTM1 gene, were at higher risk (as much as ninefold) of developing lung cancer when exposed to a relatively low level of cigarette smoke. Interestingly, there appear to be interethnic differences in the significance of variant genes which necessitate further study in order to elucidate the precise role of such alterations in susceptibility to disease (Kalow 1962; Nebert and McKinnon 1994; Kawajiri, Watanabe and Hayashi 1994).
Synergistic effect of two or more polymorphisms on the toxic response
A toxic response to an environmental agent may be greatly exaggerated by the combination of two pharmacogenetic defects in the same individual, for example, the combined effects of the N-acetyltransferase (NAT2) polymorphism and the glucose-6-phosphate dehydrogenase (G6PD) polymorphism.
Occupational exposure to arylamines constitutes a grave risk of urinary bladder cancer. Since the elegant studies of Cartwright in 1954, it has become clear that the N-acetylator status is a determinant of azo-dye-induced bladder cancer. There is a highly significant correlation between the slow-acetylator phenotype and the occurrence of bladder cancer, as well as the degree of invasiveness of this cancer in the bladder wall. On the contrary, there is a significant association between the rapid-acetylator phenotype and the incidence of colorectal carcinoma. The N-acetyltransferase (NAT1, NAT2) genes have been cloned and sequenced, and DNA-based assays are now able to detect the more than a dozen allelic variants which account for the slow-acetylator phenotype. The NAT2 gene is polymorphic and responsible for most of the variability in toxic response to environmental chemicals (Weber 1987; Grant 1993).
Glucose-6-phosphate dehydrogenase (G6PD) is an enzyme critical in the generation and maintenance of NADPH. Low or absent G6PD activity can lead to severe drug- or xenobiotic-induced haemolysis, due to the absence of normal levels of reduced glutathione (GSH) in the red blood cell. G6PD deficiency affects at least 300 million people worldwide. More than 10% of African-American males exhibit the less severe phenotype, while certain Sardinian communities exhibit the more severe “Mediterranean type” at frequencies as high as one in every three persons. The G6PD gene has been cloned and localized to the X chromosome, and numerous diverse point mutations account for the large degree of phenotypic heterogeneity seen in G6PD-deficient individuals (Beutler 1992).
Thiozalsulphone, an arylamine sulpha drug, was found to cause a bimodal distribution of haemolytic anaemia in the treated population. When treated with certain drugs, individuals with the combination of G6PD deficiency plus the slow-acetylator phenotype are more affected than those with the G6PD deficiency alone or the slow-acetylator phenotype alone. G6PD-deficient slow acetylators are at least 40 times more susceptible than normal-G6PD rapid acetylators to thiozalsulphone-induced haemolysis.
Effect of genetic polymorphisms on exposure assessment
Exposure assessment and biomonitoring (figure 1) also requires information on the genetic make-up of each individual. Given identical exposure to a hazardous chemical, the level of haemoglobin adducts (or other biomarkers) might vary by two or three orders of magnitude among individuals, depending upon each person’s metabolic fingerprint.
The same combined pharmacogenetics has been studied in chemical factory workers in Germany (table 1). Haemoglobin adducts among workers exposed to aniline and acetanilide are by far the highest in G6PD-deficient slow acetylators, as compared with the other possible combined pharmacogenetic phenotypes. This study has important implications for exposure assessment. These data demonstrate that, although two individuals might be exposed to the same ambient level of hazardous chemical in the work place, the amount of exposure (via biomarkers such as haemoglobin adducts) might be estimated to be two or more orders of magnitude less, due to the underlying genetic predisposition of the individual. Likewise, the resulting risk of an adverse health effect may vary by two or more orders of magnitude.
Table 1: Haemoglobin adducts in workers exposed to aniline and acetanilide
| Acetylator status | G6PD deficiency | |||
| Fast | Slow | No | Yes | Hgb adducts |
| + | + | 2 | ||
| + | + | 30 | ||
| + | + | 20 | ||
| + | + | 100 | ||
Source: Adapted from Lewalter and Korallus 1985.
Genetic differences in binding as well as metabolism
It should be emphasized that the same case made here for meta-bolism can also be made for binding. Heritable differences in the binding of environmental agents will greatly affect the toxic response. For example, differences in the mouse cdm gene can profoundly affect individual sensitivity to cadmium-induced testicular necrosis (Taylor, Heiniger and Meier 1973). Differences in the binding affinity of the Ah receptor are likely affect dioxin-induced toxicity and cancer (Nebert, Petersen and Puga 1991; Nebert, Puga and Vasiliou 1993).
Figure 5 summarizes the role of metabolism and binding in toxicity and cancer. Toxic agents, as they exist in the environment or following metabolism or binding, elicit their effects by either a genotoxic pathway (in which damage to DNA occurs) or a non-genotoxic pathway (in which DNA damage and mutagenesis need not occur). Interestingly, it has recently become clear that “classical” DNA-damaging agents can operate via a reduced glutathione (GSH)-dependent nongenotoxic signal transduction pathway, which is initiated on or near the cell surface in the absence of DNA and outside the cell nucleus (Devary et al. 1993). Genetic differences in metabolism and binding remain, however, as the major determinants in controlling different individual toxic responses.
Figure 5. The general means by which toxicity occurs

Role of Drug-metabolizing Enzymesin Cellular Function
Genetically based variation in drug-metabolizing enzyme function is of major importance in determining individual response to chemicals. These enzymes are pivotal in determining the fate and time course of a foreign chemical following exposure.
As illustrated in figure 5, the importance of drug-metabolizing enzymes in individual susceptibility to chemical exposure may in fact present a far more complex issue than is evident from this simple discussion of xenobiotic metabolism. In other words, during the past two decades, genotoxic mechanisms (measurements of DNA adducts and protein adducts) have been greatly emphasized. However, what if nongenotoxic mechanisms are at least as important as genotoxic mechanisms in causing toxic responses?
As mentioned earlier, the physiological roles of many drug-metabolizing enzymes involved in xenobiotic metabolism have not been accurately defined. Nebert (1994) has proposed that, because of their presence on this planet for more than 3.5 billion years, drug-metabolizing enzymes were originally (and are now still primarily) responsible for regulating the cellular levels of many nonpeptide ligands important in the transcriptional activation of genes affecting growth, differentiation, apoptosis, homeostasis and neuroendocrine functions. Furthermore, the toxicity of most, if not all, environmental agents occurs by means of agonist or antagonist action on these signal transduction pathways (Nebert 1994). Based on this hypothesis, genetic variability in drug-metabolizing enzymes may have quite dramatic effects on many critical biochemical processes within the cell, thereby leading to important differences in toxic response. It is indeed possible that such a scenario may also underlie many idiosyncratic adverse reactions encountered in patients using commonly prescribed drugs.
Conclusions
The past decade has seen remarkable progress in our understanding of the genetic basis of differential response to chemicals in drugs, foods and environmental pollutants. Drug-metabolizing enzymes have a profound influence on the way humans respond to chemicals. As our awareness of drug-metabolizing enzyme multiplicity continues to evolve, we are increasingly able to make improved assessments of toxic risk for many drugs and environmental chemicals. This is perhaps most clearly illustrated in the case of the CYP2D6 cytochrome P450 enzyme. Using relatively simple DNA-based tests, it is possible to predict the likely response of any drug predominantly metabolized by this enzyme; this prediction will ensure the safer use of valuable, yet potentially toxic, medication.
The future will no doubt see an explosion in the identification of further polymorphisms (phenotypes) involving drug-metabolizing enzymes. This information will be accompanied by improved, minimally invasive DNA-based tests to identify genotypes in human populations.
Such studies should be particularly informative in evaluating the role of chemicals in the many environmental diseases of presently unknown origin. The consideration of multiple drug-metabolizing enzyme polymorphisms, in combination (e.g., table 1), is also likely to represent a particularly fertile research area. Such studies will clarify the role of chemicals in the causation of cancers. Collectively, this information should enable the formulation of increasingly individualized advice on avoidance of chemicals likely to be of individual concern. This is the field of preventive toxicology. Such advice will no doubt greatly assist all individuals in coping with the ever increasing chemical burden to which we are exposed.
" DISCLAIMER: The ILO does not take responsibility for content presented on this web portal that is presented in any language other than English, which is the language used for the initial production and peer-review of original content. Certain statistics have not been updated since the production of the 4th edition of the Encyclopaedia (1998)."