- You are here:
-
Home

-
Contents
-
Part I. The Body
- Respiratory System

10. Respiratory System
Chapters Editors: Alois David and Gregory R. Wagner
Table of Contents
Tables and Figures
Structure and Function
Morton Lippmann
Lung Function Examination
Ulf Ulfvarson and Monica Dahlqvist
Diseases Caused by Respiratory Irritants and Toxic Chemicals
David L.S. Ryon and William N. Rom
Occupational Asthma
George Friedman-Jimenez and Edward L. Petsonk
Diseases Caused by Organic Dusts
Ragnar Rylander and Richard S. F. Schilling
Beryllium Disease
Homayoun Kazemi
Pneumoconioses: Definition
Alois David
ILO International Classification of Radiographs of Pneumoconioses
Michel Lesage
Aetiopathogenesis of Pneumoconioses
Patrick Sébastien and Raymond Bégin
Silicosis
John E. Parker and Gregory R. Wagner
Coal Workers’ Lung Diseases
Michael D. Attfield, Edward L. Petsonk and Gregory R. Wagner
Asbestos-Related Diseases
Margaret R. Becklake
Hard Metal Disease
Gerolamo Chiappino
Respiratory System: The Variety of Pneumoconioses
Steven R. Short and Edward L. Petsonk
Chronic Obstructive Pulmonary Disease
Kazimierz Marek and Jan E. Zejda
Health Effects of Man-Made Fibres
James E. Lockey and Clara S. Ross
Respiratory Cancer
Paolo Boffetta and Elisabete Weiderpass
Occupationally Acquired Infections of the Lung
Anthony A. Marfin, Ann F. Hubbs, Karl J. Musgrave, and John E. Parker
Tables
Click a link below to view table in article context.
1. Respiratory tract regions & particle deposition models
2. Inhalable, thoracic & respirable dust criteria
3. Summary of respiratory irritants
4. Mechanisms of lung injury by inhaled substances
5. Compounds capable of lung toxicity
6. Medical case definition of occupational asthma
7. Steps in diagnostic evaluation of asthma in the workplace
8. Sensitizing agents that can cause occupational asthma
9. Examples of sources of hazards of exposure to organic dust
10. Agents in organic dusts with potential biological activity
11. Diseases induced by organic dusts & their ICD codes
12. Diagnostic criteria for byssinosis
13. Properties of beryllium & its compounds
14. Description of standard radiographs
15. ILO 1980 Classification: Radiographs of Pneumoconioses
16. Asbestos-related diseases & conditions
17. Main commercial sources, products & uses of asbestos
18. Prevalence of COPD
19. Risk factors implicated in COPD
20. Loss of ventilatory function
21. Diagnostic classification, chronic bronchitis & emphysema
22. Lung function testing in COPD
23. Synthetic fibres
24. Established human respiratory carcinogens (IARC)
25. Probable human respiratory carcinogens (IARC)
26. Occupationally acquired respiratory infectious diseases
Figures
Point to a thumbnail to see figure caption, click to see figure in article context.
|
|























Structure and Function
The respiratory system extends from the breathing zone just outside of the nose and mouth through the conductive airways in the head and thorax to the alveoli, where respiratory gas exchange takes place between the alveoli and the capillary blood flowing around them. Its prime function is to deliver oxygen (O2) to the gas-exchange region of the lung, where it can diffuse to and through the walls of the alveoli to oxygenate the blood passing through the alveolar capillaries as needed over a wide range of work or activity levels. In addition, the system must also: (1) remove an equal volume of carbon dioxide entering the lungs from the alveolar capillaries; (2) maintain body temperature and water vapour saturation within the lung airways (in order to maintain the viability and functional capacities of the surface fluids and cells); (3) maintain sterility (to prevent infections and their adverse consequences); and (4) eliminate excess surface fluids and debris, such as inhaled particles and senescent phagocytic and epithelial cells. It must accomplish all of these demanding tasks continuously over a lifetime, and do so with high efficiency in terms of performance and energy utilization. The system can be abused and overwhelmed by severe insults such as high concentrations of cigarette smoke and industrial dust, or by low concentrations of specific pathogens which attack or destroy its defence mechanisms, or cause them to malfunction. Its ability to overcome or compensate for such insults as competently as it usually does is a testament to its elegant combination of structure and function.
Mass Transfer
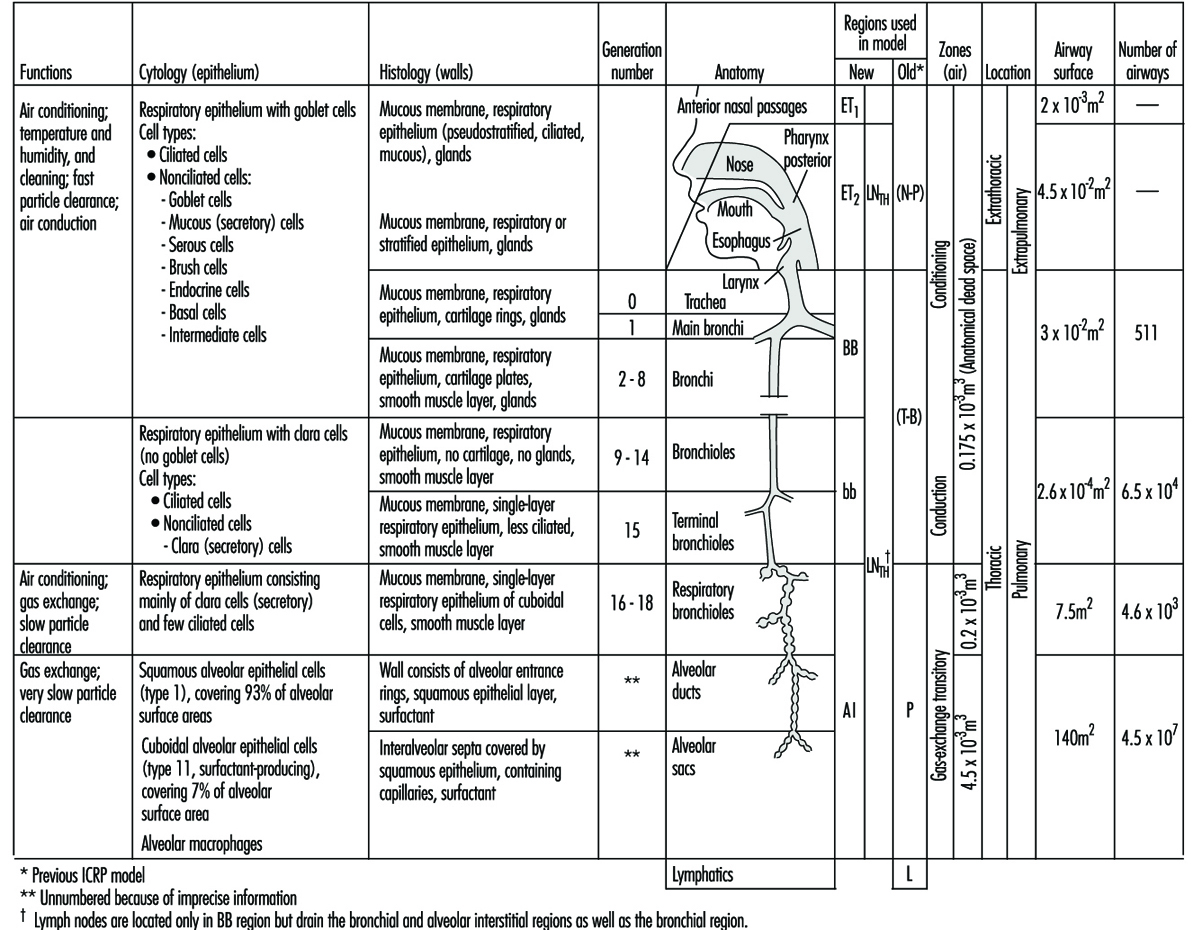
The complex structure and numerous functions of the human respiratory tract have been summarized concisely by a Task Group of the International Commission on Radiological Protection (ICRP 1994), as shown in figure 1. The conductive airways, also known as the respiratory dead space, occupy about 0.2 litres. They condition the inhaled air and distribute it, by convective (bulk) flow, to the approximately 65,000 respiratory acini leading off the terminal bronchioles. As tidal volumes increase, convective flow dominates gas exchange deeper into the respiratory bronchioles. In any case, within the respiratory acinus, the distance from the convective tidal front to alveolar surfaces is short enough so that efficient CO2-O2 exchange takes place by molecular diffusion. By contrast, airborne particles, with diffusion coefficients smaller by orders of magnitude than those for gases, tend to remain suspended in the tidal air, and can be exhaled without deposition.
Figure 1. Morphometry, cytology, histology, function and structure of the respiratory tract and regions used in the 1994 ICRP dosimetry model.
A significant fraction of the inhaled particles do deposit within the respiratory tract. The mechanisms accounting for particle deposition in the lung airways during the inspiratory phase of a tidal breath are summarized in figure 2. Particles larger than about 2 mm in aerodynamic diameter (diameter of a unit density sphere having the same terminal settling (Stokes) velocity) can have significant momentum and deposit by impaction at the relatively high velocities present in the larger airways. Particles larger than about 1 mm can deposit by sedimentation in the smaller conductive airways, where flow velocities are very low. Finally, particles with diameters between 0.1 and 1 mm, which have a very low probability of depositing during a single tidal breath, can be retained within the approximately 15% of the inspired tidal air that is exchanged with residual lung air during each tidal cycle. This volumetric exchange occurs because of the variable time-constants for airflow in the different segments of the lungs. Due to the much longer residence times of the residual air in the lungs, the low intrinsic particle displacements of 0.1 to 1 mm particles within such trapped volumes of inhaled tidal air become sufficient to cause their deposition by sedimentation and/or diffusion over the course of successive breaths.
Figure 2. Mechanisms for particle deposition in lung airways
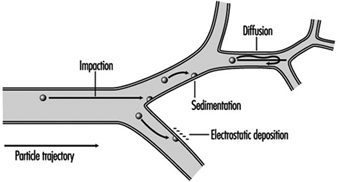
The essentially particle-free residual lung air that accounts for about 15% of the expiratory tidal flow tends to act like a clean-air sheath around the axial core of distally moving tidal air, such that particle deposition in the respiratory acinus is concentrated on interior surfaces such as airway bifurcations, while interbranch airway walls have little deposition.
The number of particles deposited and their distribution along the respiratory tract surfaces are, along with the toxic properties of the material deposited, the critical determinants of pathogenic potential. The deposited particles can damage the epithelial and/or the mobile phagocytic cells at or near the deposition site, or can stimulate the secretion of fluids and cell-derived mediators that have secondary effects on the system. Soluble materials deposited as, on, or within particles can diffuse into and through surface fluids and cells and be rapidly transported by the bloodstream throughout the body.
Aqueous solubility of bulk materials is a poor guide to particle solubility in the respiratory tract. Solubility is generally greatly enhanced by the very large surface-to-volume ratio of particles small enough to enter the lungs. Furthermore, the ionic and lipid contents of surface fluids within the airways are complex and highly variable, and can lead to either enhanced solubility or to rapid precipitation of aqueous solutes. Furthermore, the clearance pathways and residence times for particles on airway surfaces are very different in the different functional parts of the respiratory tract.
The revised ICRP Task Group’s clearance model identifies the principal clearance pathways within the respiratory tract that are important in determining the retention of various radioactive materials, and thus the radiation doses received by respiratory tissues and other organs after translocation. The ICRP deposition model is used to estimate the amount of inhaled material that enters each clearance pathway. These discrete pathways are represented by the compartment model shown in figure 3. They correspond to the anatomic compartments illustrated in Figure 1, and are summarized in table 1, along with those of other groups providing guidance on the dosimetry of inhaled particles.
Figure 3. Compartment model to represent time-dependent particle transport from each region in 1994 ICRP model
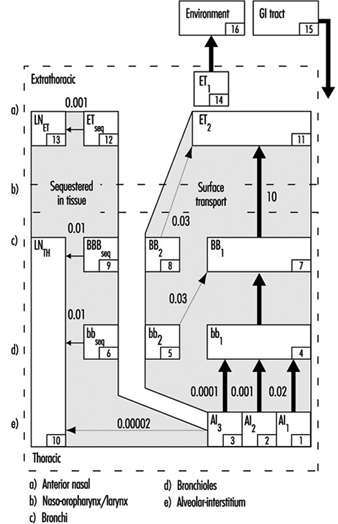
Table 1. Respiratory tract regions as defined in particle deposition models
| Anatomic structures included | ACGIH Region | ISO and CEN Regions | 1966 ICRP Task Group Region | 1994 ICRP Task Group Region |
| Nose, nasopharynx Mouth, oropharynx, laryngopharynx |
Head airways (HAR) | Extrathoracic (E) | Nasopharynx (NP) | Anterior nasal passages (ET1 ) All other extrathoracic (ET2 ) |
| Trachea, bronchi | Tracheobronchial (TBR) | Tracheobronchial (B) | Tracheobronchial (TB) | Trachea and large bronchi (BB) |
| Bronchioles (to terminal bronchioles) | Bronchioles (bb) | |||
| Respiratory bronchioles, alveolar ducts, alveolar sacs, alveoli |
Gas exchange (GER) | Alveolar (A) | Pulmonary (P) | Alveolar-interstitial (AI) |
Extrathoracic airways
As shown in figure 1, the extrathoracic airways were partitioned by ICRP (1994) into two distinct clearance and dosimetric regions: the anterior nasal passages (ET1) and all other extrathoracic airways (ET2)—that is, the posterior nasal passages, the naso- and oropharynx, and the larynx. Particles deposited on the surface of the skin lining the anterior nasal passages (ET1) are assumed to be subject only to removal by extrinsic means (nose blowing, wiping and so on). The bulk of material deposited in the naso-oropharynx or larynx (ET2) is subject to fast clearance in the layer of fluid that covers these airways. The new model recognizes that diffusional deposition of ultrafine particles in the extrathoracic airways can be substantial, while the earlier models did not.
Thoracic airways
Radioactive material deposited in the thorax is generally divided between the tracheobronchial (TB) region, where deposited particles are subject to relatively fast mucociliary clearance, and the alveolar-interstitial (AI) region, where the particle clearance is much slower.
For dosimetry purposes, the ICRP (1994) divided deposition of inhaled material in the TB region between the trachea and bronchi (BB), and the more distal, small airways, the bronchioles (bb). However, the subsequent efficiency with which cilia in either type of airways are able to clear deposited particles is controversial. In order to be certain that doses to bronchial and bronchiolar epithelia would not be underestimated, the Task Group assumed that as much as half the number of particles deposited in these airways is subject to relatively “slow” mucociliary clearance. The likelihood that a particle is cleared relatively slowly by the mucociliary system appears to depend on its physical size.
Material deposited in the AI region is subdivided among three compartments (AI1, AI2 and AI3) that are each cleared more slowly than TB deposition, with the subregions cleared at different characteristic rates.
Figure 4. Fractional deposition in each region of respiratory tract for reference light worker (normal nose breather) in 1994 ICRP model.
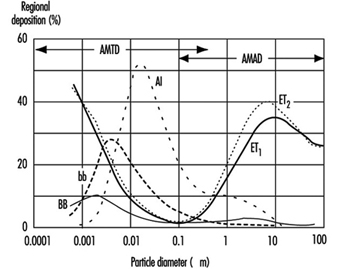
Figure 4 depicts the predictions of the ICRP (1994) model in terms of the fractional deposition in each region as a function of the size of the inhaled particles. It reflects the minimal lung deposition between 0.1 and 1 mm, where deposition is determined largely by the exchange, in the deep lung, between tidal and residual lung air. Deposition increases below 0.1 mm as diffusion becomes more efficient with decreasing particle size. Deposition increases with increasing particle size above 1 mm as sedimentation and impaction become increasingly effective.
Less complex models for size-selective deposition have been adopted by occupational health and community air pollution professionals and agencies, and these have been used to develop inhalation exposure limits within specific particle size ranges. Distinctions are made between:
- those particles that are not aspirated into the nose or mouth and therefore represent no inhalation hazard
- the inhalable (also known as inspirable) particulate mass (IPM)—those that are inhaled and are hazardous when deposited anywhere within the respiratory tract
- the thoracic particulate mass (TPM)—those that penetrate the larynx and are hazardous when deposited anywhere within the thorax and
- the respirable particulate mass (RPM)—those particles that penetrate through the terminal bronchioles and are hazardous when deposited within the gas-exchange region of the lungs.
In the early 1990s there has been an international harmonization of the quantitative definitions of IPM, TPM and RPM. The size-selective inlet specifications for air samplers meeting the criteria of the American Conference of Governmental Industrial Hygienists (ACGIH 1993), the International Organization for Standardization (ISO 1991) and the European Standardization Committee (CEN 1991) are enumerated in table 2. They differ from the deposition fractions of ICRP (1994), especially for larger particles, because they take the conservative position that protection should be provided for those engaged in oral inhalation, and thereby bypass the more efficient filtration efficiency of the nasal passages.
Table 2. Inhalable, thoracic and respirable dust criteria of ACGIH, ISO and CEN, and PM10 criteria of US EPA
| Inhalable | Thoracic | Respirable | PM10 | ||||
| Particle aero- dynamic diameter (mm) |
Inhalable Particulate Mass (IPM) (%) |
Particle aero- dynamic diameter (mm) |
Thoracic Particulate Mass (TPM) (%) |
Particle aero- dynamic diameter (mm) |
Respirable Particulate Mass (RPM) (%) |
Particle aero- dynamic diameter (mm) |
Thoracic Particulate Mass (TPM) (%) |
| 0 | 100 | 0 | 100 | 0 | 100 | 0 | 100 |
| 1 | 97 | 2 | 94 | 1 | 97 | 2 | 94 |
| 2 | 94 | 4 | 89 | 2 | 91 | 4 | 89 |
| 5 | 87 | 6 | 80.5 | 3 | 74 | 6 | 81.2 |
| 10 | 77 | 8 | 67 | 4 | 50 | 8 | 69.7 |
| 20 | 65 | 10 | 50 | 5 | 30 | 10 | 55.1 |
| 30 | 58 | 12 | 35 | 6 | 17 | 12 | 37.1 |
| 40 | 54.5 | 14 | 23 | 7 | 9 | 14 | 15.9 |
| 50 | 52.5 | 16 | 15 | 8 | 5 | 16 | 0 |
| 100 | 50 | 18 | 9.5 | 10 | 1 | ||
| 20 | 6 | ||||||
| 25 | 2 | ||||||
The US Environmental Protection Agency (EPA 1987) standard for ambient air particle concentration is known as PM10, that is, particulate matter less than 10 mm in aerodynamic diameter. It has a sampler inlet criterion that is similar (functionally equivalent) to TPM but, as shown in Table 2, somewhat different numerical specifications.
Air Pollutants
Pollutants can be dispersed in air at normal ambient temperatures and pressures in gaseous, liquid and solid forms. The latter two represent suspensions of particles in air and were given the generic term aerosols by Gibbs (1924) on the basis of analogy to the term hydrosol, used to describe dispersed systems in water. Gases and vapours, which are present as discrete molecules, form true solutions in air. Particles consisting of moderate to high vapour pressure materials tend to evaporate rapidly, because those small enough to remain suspended in air for more than a few minutes (i.e., those smaller than about 10 mm) have large surface-to-volume ratios. Some materials with relatively low vapour pressures can have appreciable fractions in both vapour and aerosol forms simultaneously.
Gases and vapours
Once dispersed in air, contaminant gases and vapours generally form mixtures so dilute that their physical properties (such as density, viscosity, enthalpy and so on) are indistinguishable from those of clean air. Such mixtures may be considered to follow ideal gas law relationships. There is no practical difference between a gas and a vapour except that the latter is generally considered to be the gaseous phase of a substance that can exist as a solid or liquid at room temperature. While dispersed in air, all molecules of a given compound are essentially equivalent in their size and probabilities of capture by ambient surfaces, respiratory tract surfaces and contaminant collectors or samplers.
Aerosols
Aerosols, being dispersions of solid or liquid particles in air, have the very significant additional variable of particle size. Size affects particle motion and, hence, the probabilities of physical phenomena such as coagulation, dispersion, sedimentation, impaction onto surfaces, interfacial phenomena and light-scattering properties. It is not possible to characterize a given particle by a single size parameter. For example, a particle’s aerodynamic properties depend on density and shape as well as linear dimensions, and the effective size for light scattering is dependent on refractive index and shape.
In some special cases, all of the particles are essentially the same in size. Such aerosols are considered to be monodisperse. Examples are natural pollens and some laboratory-generated aerosols. More typically, aerosols are composed of particles of many different sizes and hence are called heterodisperse or polydisperse. Different aerosols have different degrees of size dispersion. It is, therefore, necessary to specify at least two parameters in characterizing aerosol size: a measure of central tendency, such as a mean or median, and a measure of dispersion, such as an arithmetic or geometric standard deviation.
Particles generated by a single source or process generally have diameters following a log-normal distribution; that is, the logarithms of their individual diameters have a Gaussian distribution. In this case, the measure of dispersion is the geometric standard deviation, which is the ratio of the 84.1 percentile size to the 50 percentile size. When more than one source of particles is significant, the resulting mixed aerosol will usually not follow a single log-normal distribution, and it may be necessary to describe it by the sum of several distributions.
Particle characteristics
There are many properties of particles other than their linear size that can greatly influence their airborne behaviour and their effects on the environment and health. These include:
Surface. For spherical particles, the surface varies as the square of the diameter. However, for an aerosol of given mass concentration, the total aerosol surface increases with decreasing particle size. For non-spherical or aggregate particles, and for particles with internal cracks or pores, the ratio of surface to volume can be much greater than for spheres.
Volume. Particle volume varies as the cube of the diameter; therefore, the few largest particles in an aerosol tend to dominate its volume (or mass) concentration.
Shape. A particle’s shape affects its aerodynamic drag as well as its surface area and therefore its motion and deposition probabilities.
Density. A particle’s velocity in response to gravitational or inertial forces increases as the square root of its density.
Aerodynamic diameter. The diameter of a unit-density sphere having the same terminal settling velocity as the particle under consideration is equal to its aerodynamic diameter. Terminal settling velocity is the equilibrium velocity of a particle that is falling under the influence of gravity and fluid resistance. Aerodynamic diameter is determined by the actual particle size, the particle density and an aerodynamic shape factor.
Types of aerosols
Aerosols are generally classified in terms of their processes of formation. Although the following classification is neither precise nor comprehensive, it is commonly used and accepted in the industrial hygiene and air pollution fields.
Dust. An aerosol formed by mechanical subdivision of bulk material into airborne fines having the same chemical composition. Dust particles are generally solid and irregular in shape and have diameters greater than 1 mm.
Fume. An aerosol of solid particles formed by condensation of vapours formed by combustion or sublimation at elevated temperatures. The primary particles are generally very small (less than 0.1 mm) and have spherical or characteristic crystalline shapes. They may be chemically identical to the parent material, or may be composed of an oxidation product such as metal oxide. Since they may be formed in high number concentrations, they often rapidly coagulate, forming aggregate clusters of low overall density.
Smoke. An aerosol formed by condensation of combustion products, generally of organic materials. The particles are generally liquid droplets with diameters less than 0.5 mm.
Mist. A droplet aerosol formed by mechanical shearing of a bulk liquid, for example, by atomization, nebulization, bubbling or spraying. The droplet size can cover a very large range, usually from about 2 mm to greater than 50 mm.
Fog. An aqueous aerosol formed by condensation of water vapour on atmospheric nuclei at high relative humidities. The droplet sizes are generally greater than 1 mm.
Smog. A popular term for a pollution aerosol derived from a combination of smoke and fog. It is now commonly used for any atmospheric pollution mixture.
Haze. A submicrometer-sized aerosol of hygroscopic particles that take up water vapour at relatively low relative humidities.
Aitken or condensation nuclei (CN). Very small atmospheric particles (mostly smaller than 0.1 mm) formed by combustion processes and by chemical conversion from gaseous precursors.
Accumulation mode. A term given to the particles in the ambient atmosphere ranging from 0.1 to about 1.0 mm in diameter. These particles generally are spherical (having liquid surfaces), and form by coagulation and condensation of smaller particles that derive from gaseous precursors. Being too large for rapid coagulation and too small for effective sedimentation, they tend to accumulate in the ambient air.
Coarse particle mode. Ambient air particles larger than about 2.5 mm in aerodynamic diameter and generally formed by mechanical processes and surface dust resuspension.
Biological Responses of the Respiratory System to Air Pollutants
Responses to air pollutants range from nuisance to tissue necrosis and death, from generalized systemic effects to highly specific attacks on single tissues. Host and environmental factors serve to modify the effects of inhaled chemicals, and the ultimate response is the result of their interaction. The main host factors are:
- age—for example, older people, especially those with chronically reduced cardiovascular and respiratory function, who may not be able to cope with additional pulmonary stresses
- state of health—for example, concurrent disease or dysfunction
- nutritional status
- immunological status
- sex and other genetic factors—for example, enzyme-related differences in biotransformation mechanisms, such as deficient metabolic pathways, and inability to synthesize certain detoxification enzymes
- psychological state—for example, stress, anxiety and
- cultural factors—for example, cigarette smoking, which may affect normal defences, or may potentiate the effect of other chemicals.
The environmental factors include the concentration, stability and physicochemical properties of the agent in the exposure environment and the duration, frequency and route of exposure. Acute and chronic exposures to a chemical may result in different pathological manifestations.
Any organ can respond in only a limited number of ways, and there are numerous diagnostic labels for the resultant diseases. The following sections discuss the broad types of responses of the respiratory system which may occur following exposure to environmental pollutants.
Irritant response
Irritants produce a pattern of generalized, non-specific tissue inflammation, and destruction may result at the area of contaminant contact. Some irritants produce no systemic effect because the irritant response is much greater than any systemic effect, while some also have significant systemic effects following absorption—for example, hydrogen sulphide absorbed via the lungs.
At high concentrations, irritants may cause a burning sensation in the nose and throat (and usually also in the eyes), pain in the chest and coughing producing inflammation of the mucosa (tracheitis, bronchitis). Examples of irritants are gases such as chlorine, fluorine, sulphur dioxide, phosgene and oxides of nitrogen; mists of acids or alkali; fumes of cadmium; dusts of zinc chloride and vanadium pentoxide. High concentrations of chemical irritants may also penetrate deep into the lungs and cause lung oedema (the alveoli are filled with liquid) or inflammation (chemical pneumonitis).
Highly elevated concentrations of dusts which have no chemical irritative properties can also mechanically irritate bronchi and, after entering the gastrointestinal tract, may also contribute to stomach and colon cancer.
Exposure to irritants may result in death if critical organs are severely damaged. On the other hand, the damage may be reversible, or it may result in permanent loss of some degree of function, such as impaired gas-exchange capacity.
Fibrotic response
A number of dusts lead to the development of a group of chronic lung disorders termed pneumoconioses. This general term encompasses many fibrotic conditions of the lung, that is, diseases characterized by scar formation in the interstitial connective tissue. Pneumoconioses are due to the inhalation and subsequent selective retention of certain dusts in the alveoli, from which they are subject to interstitial sequestration.
Pneumoconioses are characterized by specific fibrotic lesions, which differ in type and pattern according to the dust involved. For example, silicosis, due to the deposition of crystalline-free silica, is characterized by a nodular type of fibrosis, while a diffuse fibrosis is found in asbestosis, due to asbestos-fibre exposure. Certain dusts, such as iron oxide, produce only altered radiology (siderosis) with no functional impairment, while the effects of others range from a minimal disability to death.
Allergic response
Allergic responses involve the phenomenon known as sensitization. Initial exposure to an allergen results in the induction of antibody formation; subsequent exposure of the now “sensitized” individual results in an immune response—that is, an antibody-antigen reaction (the antigen is the allergen in combination with an endogenous protein). This immune reaction may occur immediately following exposure to the allergen, or it may be a delayed response.
The primary respiratory allergic reactions are bronchial asthma, reactions in the upper respiratory tract which involve the release of histamine or histamine-like mediators following immune reactions in the mucosa, and a type of pneumonitis (lung inflammation) known as extrinsic allergic alveolitis. In addition to these local reactions, a systemic allergic reaction (anaphylactic shock) may follow exposure to some chemical allergens.
Infectious response
Infectious agents can cause tuberculosis, anthrax, ornithosis, brucellosis, histoplasmosis, Legionnaires’ disease and so on.
Carcinogenic response
Cancer is a general term for a group of related diseases characterized by the uncontrolled growth of tissues. Its development is due to a complex process of interacting multiple factors in the host and the environment.
One of the great difficulties in attempting to relate exposure to a specific agent to cancer development in humans is the long latent period, typically from 15 to 40 years, between onset of exposure and disease manifestation.
Examples of air pollutants that can produce cancer of the lungs are arsenic and its compounds, chromates, silica, particles containing polycyclic aromatic hydrocarbons and certain nickel-bearing dusts. Asbestos fibres can cause bronchial cancer and mesothelioma of the pleura and peritoneum. Deposited radioactive particles may expose lung tissue to high local doses of ionizing radiation and be the cause of cancer.
Systemic response
Many environmental chemicals produce a generalized systemic disease due to their effects upon a number of target sites. Lungs are not only the target for many harmful agents but the site of entry of toxic substances which pass through the lungs into the bloodstream without any damage to the lungs. However, when distributed by the blood circulation to various organs, they can damage them or cause general poisoning and have systemic effects. This role of the lungs in occupational pathology is not the subject of this article. However, the effect of finely dispersed particulates (fumes) of several metal oxides which are often associated with an acute systemic syndrome known as metal fume fever should be mentioned.
Lung Function Examination
Lung function may be measured in a number of ways. However, the aim of the measurements has to be clear before the examination, in order to interpret the results correctly. In this article we will discuss lung function examination with special regard to the occupational field. It is important to remember the limitations in different lung function measurements. Acute temporary lung function effects may not be discernible in case of exposure to fibrogenic dust like quartz and asbestos, but chronic effects on lung function after long-term (>20 years) exposure may be. This is due to the fact that chronic effects occur years after the dust is inhaled and deposited in the lungs. On the other hand, acute temporary effects of organic and inorganic dust, as well as mould, welding fumes and motor exhaust, are well suited to study. This is due to the fact that the irritative effect of these dusts will occur after a few hours of exposure. Acute or chronic lung function effects also may be discernible in cases of exposure to concentrations of irritating gases (nitrogen dioxide, aldehydes, acids and acid chlorides) in the vicinity of well documented exposure limit values, especially if the effect is potentiated by particulate air contamination.
Lung function measurements have to be safe for the examined subjects, and the lung function equipment has to be safe for the examiner. A summary of the specific requirements for different kinds of lung function equipment are available (e.g., Quanjer et al. 1993). Of course, the equipment must be calibrated according to independent standards. This may be difficult to achieve, especially when computerized equipment is being used. The result of the lung function test is dependent on both the subject and the examiner. To provide satisfactory results from the examination, technicians have to be well trained, and able to instruct the subject carefully and also encourage the subject to carry out the test properly. The examiner should also have knowledge about the airways and lungs in order to interpret the results from the recordings correctly.
It is recommended that the methods used have a fairly high reproducibility both between and within subjects. Reproducibility may be measured as the coefficient of variation, that is, the standard deviation multiplied by 100 divided by the mean value. Values below 10% in repeated measurements on the same subject are deemed acceptable.
In order to determine if the measured values are pathological or not, they must be compared with prediction equations. Usually the prediction equations for spirometric variables are based on age and height, stratified for sex. Men have on the average higher lung function values than women, of the same age and height. Lung function decreases with age and increases with height. A tall subject will therefore have higher lung volume than a short subject of the same age. The outcome from prediction equations may differ considerably between different reference populations. The variation in age and height in the reference population will also influence the predicted values. This means, for example, that a prediction equation must not be used if age and/or height for the examined subject are outside the ranges for the population that is the basis for the prediction equation.
Smoking will also diminish lung function, and the effect may be potentiated in subjects who are occupationally exposed to irritating agents. Lung function used to be considered as not being pathological if the obtained values are within 80% of the predicted value, derived from a prediction equation.
Measurements
Lung function measurements are carried out to judge the condition of the lungs. Measurements may either concern single or multiple measured lung volumes, or the dynamic properties in the airways and lungs. The latter is usually determined through effort-dependent manoeuvres. The conditions in the lungs may also be examined with regard to their physiological function, that is, diffusion capacity, airway resistance and compliance (see below).
Measurements concerning ventilatory capacity are obtained by spirometry. The breathing manoeuvre is usually performed as a maximal inspiration followed by a maximal expiration, vital capacity (VC, measured in litres). At least three technically satisfactory recordings (i.e., full inspiration and expiration effort and no observed leaks) should be done, and the highest value reported. The volume may be directly measured by a water-sealed or a low-resistive bell, or indirectly measured by pneumotachography (i.e., integration of a flow signal over time). It is important here to note that all measured lung volumes should be expressed in BTPS, that is, body temperature and ambient pressure saturated with water vapour.
Forced expired vital capacity (FVC, in litres) is defined as a VC measurement performed with a maximally forced expiratory effort. Due to the simplicity of the test and the relatively inexpensive equipment, the forced expirogram has become a useful test in the monitoring of lung function. However, this has resulted in many poor recordings, of which the practical value is debatable. In order to carry out satisfactory recordings, the updated guideline for the collection and use of the forced expirogram, published by the American Thoracic Society in 1987, may be useful.
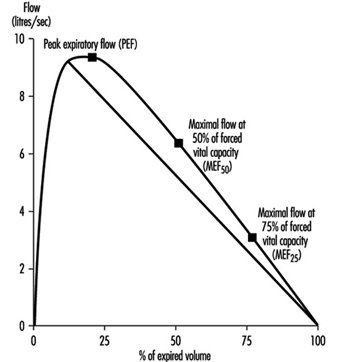
Instantaneous flows may be measured on flow-volume or flow-time curves, while time average flows or times are derived from the spirogram. Associated variables which can be calculated from the forced expirogram are forced expired volume in one second (FEV1, in litres per second), in percentage of FVC (FEV1%), peak flow (PEF, l/s), maximal flows at 50% and 75% of forced vital capacity (MEF50 and MEF25, respectively). An illustration of the derivation of FEV1 from the forced expirogram is outlined in figure 1. In healthy subjects, maximal flow rates at large lung volumes (i.e., at the beginning of expiration) reflect mainly the flow characteristics of the large airways while those at small lung volumes (i.e., the end of expiration) are usually held to reflect the characteristics of the small airways, figure 2. In the latter the flow is laminar, while in the large airways it may be turbulent.
Figure 1. Forced expiratory spirogram showing the derivation of FEV1 and FVC according to the extrapolation principle.
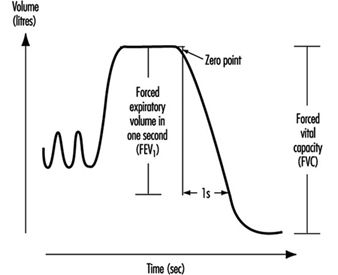
Figure 2. Flow-volume curve showing the derivation of peak expiratory flow (PEF), maximal flows at 50% and 75% of forced vital capacity (![]() and
and ![]() , respectively).
, respectively).
PEF may also be measured by a small portable device such as the one developed by Wright in 1959. An advantage with this equipment is that the subject may carry out serial measurements—for example, at the workplace. To get useful recordings, however, it is necessary to instruct the subjects well. Moreover, one should keep in mind that measurements of PEF with, for example, a Wright meter and those measured by conventional spirometry should not be compared due to the different blow techniques.
The spirometric variables VC, FVC and FEV1 show a reasonable variation between individuals where age, height and sex usually explain 60 to 70% of the variation. Restrictive lung function disorders will result in lower values for VC, FVC and FEV1. Measurements of flows during expiration show a great individual variation, since the measured flows are both effort and time dependent. This means, for example, that a subject will have extremely high flow in case of diminished lung volume. On the other hand, the flow may be extremely low in case of very high lung volume. However, the flow is usually decreased in case of a chronic obstructive disease (e.g., asthma, chronic bronchitis).
Figure 3. A principal outline of the equipment for determination of total lung capacity (TLC) according to the helium dilution technique.
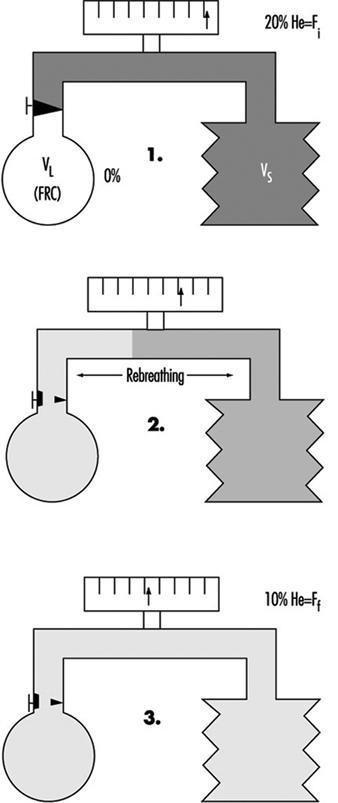
The proportion of residual volume (RV), that is, the volume of air which still is in the lungs after a maximal expiration, can be determined by gas dilution or by body plethysmography. The gas dilution technique requires less complicated equipment and is therefore more convenient to use in studies carried out at the workplace. In figure 3, the principle for the gas dilution technique has been outlined. The technique is based on dilution of an indicator gas in a rebreathing circuit. The indicator gas must be sparingly soluble in biological tissues so that it is not taken up by the tissues and blood in the lung. Hydrogen was initially used, but because of its ability to form explosive mixtures with air it was replaced by helium, which is easily detected by means of the thermal conductivity principle.
The subject and the apparatus form a closed system, and the initial concentration of the gas is thus reduced when it is diluted into the gas volume in the lungs. After equilibration, the concentration of indicator gas is the same in the lungs as in the apparatus, and functional residual capacity (FRC) can be calculated by means of a simple dilution equation. The volume of the spirometer (including the addition of the gas mixture into the spirometer) is denoted by VS, VL is the volume of the lung, Fi is the initial gas concentration and Ff is the final concentration.
FRC = VL = [(VS · Fi) / Ff] – VS
Two to three VC manoeuvres are carried out to provide a reliable base for the calculation of TLC (in litres). The subdivisions of the different lung volumes are outlined in figure 4.
Figure 4. Spirogram labelled to show the subdivisions of the total capacity.
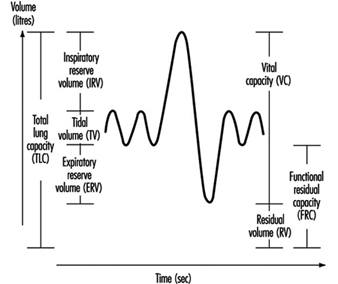
Due to change in the elastic properties of the airways, RV and FRC increase with age. In chronic obstructive diseases, increased values of RV and FRC are usually observed, while VC is decreased. However, in subjects with badly ventilated lung areas—for example, subjects with emphysema—the gas dilution technique may underestimate RV, FRC and also TLC. This is due to the fact that the indicator gas will not communicate with closed-off airways, and therefore the decrease in the indicator gas concentration will give erroneously small values.
Figure 5. A principal outline of the recording of airway closure and the slope of the alveolar plateau (%![]() ).
).
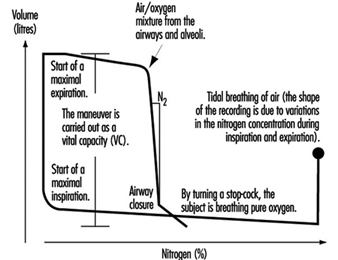
Measures of airway closure and gas distribution in the lungs can be obtained in one and the same manoeuvre by the single breath wash-out technique, figure 5. The equipment consists of a spirometer connected to a bag-in-box system and a recorder for continuous measurements of nitrogen concentration. The manoeuvre is carried out by means of a maximal inspiration of pure oxygen from the bag. In the beginning of the expiration, the nitrogen concentration increases as a result of emptying the subject’s deadspace, containing pure oxygen. The expiration continues with the air from the airways and alveoli. Finally, air from the alveoli, containing 20 to 40% nitrogen, is expired. When the expiration from the basal parts of the lungs increases, the nitrogen concentration will rise abruptly in case of airway closure in dependent lung regions, figure 5. This volume above RV, at which airways close during an expiration, is usually expressed as closing volume (CV) in percentage of VC (CV%). Distribution of the inspired air in the lungs is expressed as the slope of the alveolar plateau (%N2 or phase III, %N2/l). It is obtained by taking the difference in nitrogen concentration between the point when 30% of the air is exhaled and the point for airway closure, and dividing this by the corresponding volume.
Ageing as well as chronic obstructive disorders will result in increased values for both CV% and phase III. However, not even healthy subjects have a uniform gas distribution in the lungs, resulting in slightly elevated values for phase III, that is, 1 to 2% N2/l. The variables CV% and phase III are considered to reflect the conditions in the peripheral small airways with an internal diameter about 2 mm. Normally, the peripheral airways contribute to a small part (10 to 20%) of the total airway resistance. Quite extensive changes which are not detectable by conventional lung function tests like dynamic spirometry, may occur, for example, as a result of an exposure to irritating substances in the air in the peripheral airways. This suggests that airway obstruction begins in the small airways. Results from studies also have shown alterations in CV% and phase III before any changes from the dynamic and static spirometry have occurred. These early changes may go into remission when exposure to hazardous agents has ceased.
The transfer factor of the lung (mmol/min; kPa) is an expression of the diffusion capacity of oxygen transport into the pulmonary capillaries. The transfer factor can be determined using single or multiple breath techniques; the single breath technique is considered to be most suitable in studies at the workplace. Carbon monoxide (CO) is used since the back pressure of CO is very low in the peripheral blood, in contrast to that of oxygen. The uptake of CO is assumed to follow an exponential model, and this assumption can be used to determine the transfer factor for the lung.
Determination of TLCO (transfer factor measured with CO) is carried out by means of a breathing manoeuvre including a maximal expiration, followed by a maximal inspiration of a gas mixture containing carbon monoxide, helium, oxygen and nitrogen. After a breath-holding period, a maximal exhalation is done, reflecting the content in the alveolar air, Figure 10. Helium is used for the determination of the alveolar volume (VA). Assuming that the dilution of CO is the same as for helium, the initial concentration of CO, before the diffusion has started, can be calculated. TLCO is calculated according to the equation outlined below, where k depends on the dimensions of the component terms, t is the effective time for breath-holding and log is base 10 logarithm. Inspired volume is denoted Vi and the fractions F of CO and helium are denoted by i and a for inspired and alveolar, respectively.
TLCO = k Vi (Fa,He/Fi,He) log (Fi,CO Fa,He/Fa,CO Fi,He) (t)-1
Figure 6. A principal outline of the recording of transfer factor
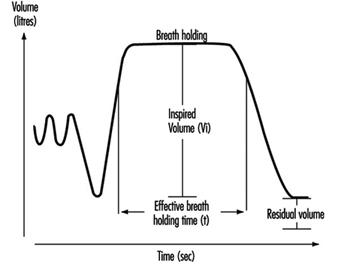
The size of TLCO will depend on a variety of conditions—for example, the amount of available haemoglobin, the volume of ventilated alveoli and perfused lung capillaries and their relation to each other. Values for TLCO decrease with age and increase with physical activity and increased lung volumes. Decreased TLCO will be found in both restrictive and obstructive lung disorders.
Compliance (l/kPa) is a function, inter alia, of the elastic property of the lungs. The lungs have an intrinsic tendency to collaborate—that is, to collapse. The power to keep the lungs stretched will depend on the elastic lung tissue, the surface tension in the alveoli, and the bronchial musculature. On the other hand, the chest wall tends to expand at lung volumes 1 to 2 litres above the FRC level. At higher lung volumes, power has to be applied to further expand the chest wall. At the FRC level, the corresponding tendency in the lungs is balanced by the tendency to expand. The FRC level is therefore denoted by the resting level of the lung.
The compliance of the lung is defined as the change in volume divided by the change in transpulmonary pressure, that is, the difference between the pressures in the mouth (atmospheric) and in the lung, as the result of a breathing manoeuvre. Measurements of the pressure in the lung are not easily carried out and are therefore replaced by measurements of the pressure in the oesophagus. The pressure in the oesophagus is almost the same as the pressure in the lung, and it is measured with a thin polyethylene catheter with a balloon covering the distal 10 cm. During inspiratory and expiratory manoeuvres, the changes in volume and pressure are recorded by means of a spirometer and pressure transducer, respectively. When the measurements are performed during tidal breathing, dynamic compliance can be measured. Static compliance is obtained when a slow VC manoeuvre is carried out. In the latter case, the measurements are carried out in a body plethysmograph, and the expiration is intermittently interrupted by means of a shutter. However, measurements of compliance are cumbersome to perform when examining exposure effects on lung function at the worksite, and this technique is considered to be more appropriate in the laboratory.
A decreased compliance (increased elasticity) is observed in fibrosis. To cause a change in volume, large changes in pressure are required. On the other hand, a high compliance is observed, for example, in emphysema as the result of loss of elastic tissue and therefore also elasticity in the lung.
The resistance in the airways essentially depends on the radius and length of the airways but also on air viscosity. The airway resistance (RL in (kPa/l) /s), can be determined by use of a spirometer, pressure transducer and a pneumotachograph (to measure the flow). The measurements may also be carried out using a body plethysmograph to record the changes in flow and pressure during panting manoeuvres. By administration of a drug intended to cause broncho-constriction, sensitive subjects, as a result of their hyperreactive airways, may be identified. Subjects with asthma usually have increased values for RL.
Acute and Chronic Effects of Occupational Exposure on Pulmonary Function
Lung function measurement may be used to disclose an occupational exposure effect on the lungs. Pre-employment examination of lung function should not be used to exclude job-seeking subjects. This is because the lung function of healthy subjects varies within wide limits and it is difficult to draw a borderline below which it can safely be stated that the lung is pathological. Another reason is that the work environment should be good enough to allow even subjects with slight lung function impairment to work safely.
Chronic effects on the lungs in occupationally exposed subjects may be detected in a number of ways. The techniques are designed to determine historical effects, however, and are less suitable to serve as guidelines to prevent lung function impairment. A common study design is to compare the actual values in exposed subjects with the lung function values obtained in a reference population without occupational exposure. The reference subjects may be recruited from the same (or nearby) workplaces or from the same city.
Multivariate analysis has been used in some studies to assess differences between exposed subjects and matched unexposed referents. Lung function values in exposed subjects may also be standardized by means of a reference equation based on lung function values in the unexposed subjects.
Another approach is to study the difference between the lung function values in exposed and unexposed workers after adjustment for age and height with the use of external reference values, calculated by means of a prediction equation based on healthy subjects. The reference population may also be matched to the exposed subjects according to ethnic group, sex, age, height and smoking habits in order to further control for those influencing factors.
The problem is, however, to decide if a decrease is large enough to be classified as pathological, when external reference values are being used. Although the instruments in the studies have to be portable and simple, attention must be paid both to the sensitivity of the chosen method for detecting small anomalies in airways and lungs and the possibility of combining different methods. There are indications that subjects with respiratory symptoms, such as exertion dyspnoea, are at a higher risk of having an accelerated decline in lung function. This means that the presence of respiratory symptoms is important and so should not be neglected.
The subject may also be followed-up by spirometry, for example, once a year, for a number of years, in order to give a warning against the development of illness. There are limitations, however, since this will be very time-consuming and the lung function may have deteriorated permanently when the decrease can be observed. This approach therefore must not be an excuse for delay in carrying out measures in order to decrease harmful concentrations of air pollutants.
Finally, chronic effects on lung function may also be studied by examining the individual changes in lung function in exposed and unexposed subjects over a number of years. One advantage of the longitudinal study design is that the intersubject variability is eliminated; however, the design is considered to be time-consuming and expensive.
Susceptible subjects may also be identified by comparing their lung function with and without exposure during working shifts. In order to minimize possible effects of diurnal variations, lung function is measured at the same time of day on one unexposed and one exposed occasion. The unexposed condition can be obtained, for example, by occasionally moving the worker to an uncontaminated area or by use of a suitable respirator during a whole shift, or in some cases by performing lung function measurements in the afternoon of a worker’s day off.
One special concern is that repeated, temporary effects can result in chronic effects. An acute temporary lung function decrease may not only be a biological exposure indicator but also a predictor of a chronic lung function decrement. Exposure to air pollutants may result in discernible acute effects on lung function, although the mean values of the measured air pollutants are below the hygienic limit values. The question thus arises, whether these effects really are harmful in the long run. This question is hard to answer directly, especially since the air pollution in workplaces often has a complex composition and the exposure cannot be described in terms of mean concentrations of single compounds. The effect of an occupational exposure is also partly due to the sensitivity of the individual. This means that some subjects will react sooner or to a larger extent than others. The underlying pathophysiological ground for an acute, temporary decrease in lung function is not fully understood. The adverse reaction upon exposure to an irritating air contaminant is, however, an objective measurement, in contrast to subjective experiences like symptoms of different origin.
The advantage of detecting early changes in airways and lungs caused by hazardous air pollutants is obvious—the prevailing exposure may be reduced in order to prevent more severe illnesses. Therefore, an important aim in this respect is to use the measurements of acute temporary effects on lung function as a sensitive early warning system that can be used when studying groups of healthy working people.
Monitoring of Irritants
Irritation is one of the most frequent criteria for setting exposure limit values. It is, however, not certain that compliance with an exposure limit based on irritation will protect against irritation. It should be considered that an exposure limit for an air contaminant usually contains at least two parts—a time-weighted average limit (TWAL) and a short-term exposure limit (STEL), or at least rules for exceeding the time-weighted average limit, “excursion limits”. In the case of highly irritating substances, such as sulphur dioxide, acrolein and phosgene, it is important to limit the concentration even during very short periods, and it has therefore been common practice to fix occupational exposure limit values in the form of ceiling limits, with a sampling period that is kept as short as the measuring facilities will allow.
Time-weighted average limit values for an eight-hour day combined with rules for excursion above these values are given for most of the substances in the American Conference of Governmental Industrial Hygienists (ACGIH) threshold limit value (TLV) list. The TLV list of 1993-94 contains the following statement concerning excursion limits for exceeding limit values:
“For the vast majority of substances with a TLV-TWA, there is not enough toxicological data available to warrant a STEL = short-term exposure limit). Nevertheless, excursions above the TLV-TWA should be controlled even where the eight-hour TWA is within recommended limits.”
Exposure measurements of known air contaminants and comparison with well documented exposure limit values should be carried out on a routine basis. There are, however, many situations when the determination of compliance with exposure limit values is not enough. This is the case in the following circumstances (inter alia):
- when the limit value is too high to safeguard against irritation
- when the irritant is unknown
- when the irritant is a complex mixture and there is no suitable indicator known.
As advocated above, the measurement of acute, temporary effects on lung function can be used in these cases as a warning against over-exposure to irritants.
In cases (2) and (3), acute, temporary effects on lung function may be applicable also in testing the efficiency of control measures to decrease exposure to air contamination or in scientific investigations, for example, in attributing biological effects to components of air contaminants. A number of examples follow in which acute, temporary lung function effects have been successfully employed in occupational health investigations.
Studies of Acute, Temporary Lung Function Effects
Work-related, temporary decrease of lung function over a work shift was recorded in cotton workers at the end of 1950. Later, several authors reported work-related, acute, temporary changes of lung function in hemp and textile workers, coal miners, workers exposed to toluene di-isocyanate, fire-fighters, rubber processing workers, moulders and coremakers, welders, ski waxers, workers exposed to organic dust and irritants in water-based paints.
However, there are also several examples where measurements before and after exposure, usually during a shift, have failed to demonstrate any acute effects, despite a high exposure. This is probably due to the effect of normal circadian variation, mainly in lung function variables depending on the size of airway calibre. Thus the temporary decrease in these variables must exceed the normal circadian variation to be recognized. The problem may be circumvented, however, by measuring lung function at the same time of the day at each study occasion. By using the exposed employee as his or her own control, the interindividual variation is further decreased. Welders were studied in this way, and although the mean difference between unexposed and exposed FVC values was less than 3% in 15 examined welders, this difference was significant at the 95% confidence level with a power of more than 99%.
The reversible transient effects on the lungs can be used as an exposure indicator of complicated irritating components. In the study cited above, particles in the work environment were crucial for the irritating effects on the airways and lungs. The particles were removed by a respirator consisting of a filter combined with a welding helmet. The results indicated that the effects on the lungs were caused by the particles in welding fumes, and that the use of a particulate respirator might prevent this effect.
Exposure to diesel exhaust also gives measurable irritative effects on the lungs, shown as an acute, temporary lung function decrease. Mechanical filters mounted on the exhaust pipes of trucks used in loading operations by stevedores relieved subjective disorders and reduced the acute, temporary lung function decrease observed when no filtration was done. The results thus indicate that the presence of particles in the work environment does play a role in the irritative effect on airways and lungs, and that it is possible to assess the effect by measurements of acute changes in lung function.
A multiplicity of exposures and a continually changing work environment may present difficulties in discerning the causal relationship of the different agents existing in a work environment. The exposure scenario in sawmills is an illuminating example. It is not possible (e.g., for economical reasons) to carry out exposure measurements of all possible agents (terpenes, dust, mould, bacteria, endotoxin, mycotoxins, etc.) in this work environment. A feasible method may be to follow the development of lung function longitudinally. In a study of sawmill workers in the wood-trimming department, lung function was examined before and after a working week, and no statistically significant decrease was found. However, a follow-up study carried out a few years later disclosed that those workers who actually had a numerical decrease in lung function during a working week also had an accelerated long-term decline in lung function. This may indicate that vulnerable subjects can be detected by measuring changes in lung function during a working week.
Diseases Caused by Respiratory Irritants and Toxic Chemicals
The presence of respiratory irritants in the workplace can be unpleasant and distracting, leading to poor morale and decreased productivity. Certain exposures are dangerous, even lethal. In either extreme, the problem of respiratory irritants and inhaled toxic chemicals is common; many workers face a daily threat of exposure. These compounds cause harm by a variety of different mechanisms, and the extent of injury can vary widely, depending on the degree of exposure and on the biochemical properties of the inhalant. However, they all have the characteristic of nonspecificity; that is, above a certain level of exposure virtually all persons experience a threat to their health.
There are other inhaled substances that cause only susceptible individuals to develop respiratory problems; such complaints are most appropriately approached as diseases of allergic and immunological origin. Certain compounds, such as isocyanates, acid anhydrides and epoxy resins, can act not only as non-specific irritants in high concentrations, but can also predispose certain subjects to allergic sensitization. These compounds provoke respiratory symptoms in sensitized individuals at very low concentrations.
Respiratory irritants include substances that cause inflammation of the airways after they are inhaled. Damage may occur in the upper and lower airways. More dangerous is acute inflammation of the pulmonary parenchyma, as in chemical pneumonitis or non-cardiogenic pulmonary oedema. Compounds that can cause parenchymal damage are considered toxic chemicals. Many inhaled toxic chemicals also act as respiratory irritants, warning us of their danger with their noxious odour and symptoms of nose and throat irritation and cough. Most respiratory irritants are also toxic to the lung parenchyma if inhaled in sufficient amount.
Many inhaled substances have systemic toxic effects after being absorbed by inhalation. Inflammatory effects on the lung may be absent, as in the case of lead, carbon monoxide or hydrogen cyanide. Minimal lung inflammation is normally seen in the inhalation fevers (e.g., organic dust toxic syndrome, metal fume fever and polymer fume fever). Severe lung and distal organ damage occurs with significant exposure to toxins such as cadmium and mercury.
The physical properties of inhaled substances predict the site of deposition; irritants will produce symptoms at these sites. Large particles (10 to 20mm) deposit in the nose and upper airways, smaller particles (5 to 10mm) deposit in the trachea and bronchi, and particles less than 5mm in size may reach the alveoli. Particles less than 0.5mm are so small they behave like gases. Toxic gases deposit according to their solubility. A water-soluble gas will be adsorbed by the moist mucosa of the upper airway; less soluble gases will deposit more randomly throughout the respiratory tract.
Respiratory Irritants
Respiratory irritants cause non-specific inflammation of the lung after being inhaled. These substances, their sources of exposure, physical and other properties, and effects on the victim are outlined in Table 1. Irritant gases tend to be more water soluble than gases more toxic to the lung parenchyma. Toxic fumes are more dangerous when they have a high irritant threshold; that is, there is little warning that the fume is being inhaled because there is little irritation.
Table 1. Summary of respiratory irritants
|
Chemical |
Sources of exposure |
Important properties |
Injury produced |
Dangerous exposure level under 15 min (PPM) |
|
Acetaldehyde |
Plastics, synthetic rubber industry, combustion products |
High vapour pressure; high water solubility |
Upper airway injury; rarely causes delayed pulmonary oedema |
|
|
Acetic acid, organic acids |
Chemical industry, electronics, combustion products |
Water soluble |
Ocular and upper airway injury |
|
|
Acid anhydrides |
Chemicals, paints, and plastics industries; components of epoxy resins |
Water soluble, highly reactive, may cause allergic sensitization |
Ocular, upper airway injury, bronchospasm; pulmonary haemorrhage after massive exposure |
|
|
Acrolein |
Plastics, textiles, pharmaceutical manufacturing, combustion products |
High vapour pressure, intermediate water solubility, extremely irritating |
Diffuse airway and parenchymal injury |
|
|
Ammonia |
Fertilizers, animal feeds, chemicals, and pharmaceuticals manufacturing |
Alkaline gas, very high water solubility |
Primarily ocular and upper airway burn; massive exposure may cause bronchiectasis |
500 |
|
Antimony trichloride, antimony penta-chloride |
Alloys, organic catalysts |
Poorly soluble, injury likely due to halide ion |
Pneumonitis, non-cardiogenic pulmonary oedema |
|
|
Beryllium |
Alloys (with copper), ceramics; electronics, aerospace and nuclear reactor equipment |
Irritant metal, also acts as an antigen to promote a long-term granulomatous response |
Acute upper airway injury, tracheobronchitis, chemical pneumonitis |
25 μg/m3 |
|
Boranes (diborane) |
Aircraft fuel, fungicide manufacturing |
Water soluble gas |
Upper airway injury, pneumonitis with massive exposure |
|
|
Hydrogen bromide |
Petroleum refining |
Upper airway injury, pneumonitis with massive exposure |
||
|
Methyl bromide |
Refrigeration, produce fumigation |
Moderately soluble gas |
Upper and lower airway injury, pneumonitis, CNS depression and seizures |
|
|
Cadmium |
Alloys with Zn and Pb, electroplating, batteries, insecticides |
Acute and chronic respiratory effects |
Tracheobronchitis, pulmonary oedema (often delayed onset over 24–48 hours); chronic low level exposure leads to inflammatory changes and emphysema |
100 |
|
Calcium oxide, calcium hydroxide |
Lime, photography, tanning, insecticides |
Moderately caustic, very high doses required for toxicity |
Upper and lower airway inflammation, pneumonitis |
|
|
Chlorine |
Bleaching, formation of chlorinated compounds, household cleaners |
Intermediate water solubilty |
Upper and lower airway inflammation, pneumonitis and non-cardiogenic pulmonary oedema |
5–10 |
|
Chloroacetophenone |
Crowd control agent, “tear gas” |
Irritant qualities are used to incapacitate; alkylating agent |
Ocular and upper airway inflammation, lower airway and parenchymal injury with masssive exposure |
1–10 |
|
o-Chlorobenzomalo- nitrile |
Crowd control agent, “tear gas” |
Irritant qualities are used to incapacitate |
Ocular and upper airway inflammation, lower airway injury with massive exposure |
|
|
Chloromethyl ethers |
Solvents, used in manufacture of other organic compounds |
Upper and lower airway irritation, also a respiratory tract carcinogen |
||
|
Chloropicrin |
Chemical manufacturing, fumigant component |
Former First World War gas |
Upper and lower airway inflammation |
15 |
|
Chromic acid (Cr(IV)) |
Welding, plating |
Water soluble irritant, allergic sensitizer |
Nasal inflammation and ulceration, rhinitis, pneumonitis with massive exposure |
|
|
Cobalt |
High temperature alloys, permanent magnets, hard metal tools (with tungsten carbide) |
Non-specific irritant, also allergic sensitizer |
Acute bronchospasm and/or pneumonitis; chronic exposure can cause lung fibrosis |
|
|
Formaldehyde |
Manufacture of foam insulation, plywood, textiles, paper, fertilizers, resins; embalming agents; combustion products |
Highly water soluble, rapidly metabolized; primarily acts via sensory nerve stimulation; sensitization reported |
Ocular and upper airway irritation; bronchospasm in severe exposure; contact dermatitis in sensitized persons |
3 |
|
Hydrochloric acid |
Metal refining, rubber manufacturing, organic compound manufacture, photographic materials |
Highly water soluble |
Ocular and upper airway inflammation, lower airway inflammation only with massive exposure |
100 |
|
Hydrofluoric acid |
Chemical catalyst, pesticides, bleaching, welding, etching |
Highly water soluble, powerful and rapid oxidant, lowers serum calcium in massive exposure |
Ocular and upper airway inflammation, tracheobronchitis and pneumonitis with massive exposure |
20 |
|
Isocyanates |
Polyurethane production; paints; herbicide and insecticide products; laminating, furniture, enamelling, resin work |
Low molecular weight organic compounds, irritants, cause sensitization in susceptible persons |
Ocular, upper and lower inflammation; asthma, hypersensitivity pneumonitis in sensitized persons |
0.1 |
|
Lithium hydride |
Alloys, ceramics, electronics, chemical catalysts |
Low solubility, highly reactive |
Pneumonitis, non-cardiogenic pulmonary oedema |
|
|
Mercury |
Electrolysis, ore and amalgam extraction, electronics manufacture |
No respiratory symptoms with low level, chronic exposure |
Ocular and respiratory tract inflammation, pneumonitis, CNS, kidney and systemic effects |
1.1 mg/m3 |
|
Nickel carbonyl |
Nickel refining, electroplating, chemical reagents |
Potent toxin |
Lower respiratory irritation, pneumonitis, delayed systemic toxic effects |
8 μg/m3 |
|
Nitrogen dioxide |
Silos after new grain storage, fertilizer making, arc welding, combustion products |
Low water solubility, brown gas at high concentration |
Ocular and upper airway inflammation, non-cardiogenic pulmonary oedema, delayed onset bronchiolitis |
50 |
|
Nitrogen mustards; sulphur mustards |
Military gases |
Causes severe injury, vesicant properties |
Ocular, upper and lower airway inflammation, pneumonitis |
20mg/m3 (N) 1 mg/m3 (S) |
|
Osmium tetroxide |
Copper refining, alloy with iridium, catalyst for steroid synthesis and ammonia formation |
Metallic osmium is inert, tetraoxide forms when heated in air |
Severe ocular and upper airway irritation; transient renal damage |
1 mg/m3 |
|
Ozone |
Arc welding, copy machines, paper bleaching |
Sweet smelling gas, moderate water solubility |
Upper and lower airway inflammation; asthmatics more susceptible |
1 |
|
Phosgene |
Pesticide and other chemical manufacture, arc welding, paint removal |
Poorly water soluble, does not irritate airways in low doses |
Upper airway inflammation and pneumonitis; delayed pulmonary oedema in low doses |
2 |
|
Phosphoric sulphides |
Production of insecticides, ignition compounds, matches |
Ocular and upper airway inflammation |
||
|
Phosphoric chlorides |
Manufacture of chlorinated organic compounds, dyes, gasoline additives |
Form phosphoric acid and hydrochloric acid on contact with mucosal surfaces |
Ocular and upper airway inflammation |
10 mg/m3 |
|
Selenium dioxide |
Copper or nickel smelting, heating of selenium alloys |
Strong vessicant, forms selenious acid (H2SeO3) on mucosal surfaces |
Ocular and upper airway inflammation, pulmonary oedema in massive exposure |
|
|
Hydrogen selenide |
Copper refining, sulphuric acid production |
Water soluble; exposure to selenium compounds gives rise to garlic odour breath |
Ocular and upper airway inflammation, delayed pulmonary oedema |
|
|
Styrene |
Manufacture of polystyrene and resins, polymers |
Highly irritating |
Ocular, upper and lower airway inflammation, neurological impairments |
600 |
|
Sulphur dioxide |
Petroleum refining, pulp mills, refrigeration plants, manufacturing of sodium sulphite |
Highly water soluble gas |
Upper airway inflammation, bronchoconstriction, pneumonitis on massive exposure |
100 |
|
Titanium tetrachloride |
Dyes, pigments, sky writing |
Chloride ions form HCl on mucosa |
Upper airway injury |
|
|
Uranium hexafluoride |
Metal coat removers, floor sealants, spray paints |
Toxicity likely from chloride ions |
Upper and lower airway injury, bronchospasm, pneumonitis |
|
|
Vanadium pentoxide |
Cleaning oil tanks, metallurgy |
Ocular, upper and lower airway symptoms |
70 |
|
|
Zinc chloride |
Smoke grenades, artillery |
More severe than zinc oxide exposure |
Upper and lower airway irritation, fever, delayed onset pneumonitis |
200 |
|
Zirconium tetrachloride |
Pigments, catalysts |
Chloride ion toxicity |
Upper and lower airway irritation, pneumonitis |
This condition is thought to result from persistent inflammation with reduction of epithelial cell layer permeability or reduced conductance threshold for subepithelial nerve endings.Adapted from Sheppard 1988; Graham 1994; Rom 1992; Blanc and Schwartz 1994; Nemery 1990; Skornik 1988.
The nature and extent of the reaction to an irritant depends on the physical properties of the gas or aerosol, the concentration and time of exposure, and on other variables as well, such as temperature, humidity and the presence of pathogens or other gases (Man and Hulbert 1988). Host factors such as age (Cabral-Anderson, Evans and Freeman 1977; Evans, Cabral-Anderson and Freeman 1977), prior exposure (Tyler, Tyler and Last 1988), level of antioxidants (McMillan and Boyd 1982) and presence of infection may play a role in determining the pathological changes seen. This wide range of factors has made it difficult to study the pathogenic effects of respiratory irritants in a systematic way.
The best understood irritants are those which inflict oxidative injury. The majority of inhaled irritants, including the major pollutants, act by oxidation or give rise to compounds that act in this way. Most metal fumes are actually oxides of the heated metal; these oxides cause oxidative injury. Oxidants damage cells primarily by lipid peroxidation, and there may be other mechanisms. On a cellular level, there is initially a fairly specific loss of ciliated cells of the airway epithelium and of Type I alveolar epithelial cells, with subsequent violation of the tight junction interface between epithelial cells (Man and Hulbert 1988; Gordon, Salano and Kleinerman 1986; Stephens et al. 1974). This leads to subepithelial and submucosal damage, with stimulation of smooth muscle and parasympathetic sensory afferent nerve endings causing bronchoconstriction (Holgate, Beasley and Twentyman 1987; Boucher 1981). An inflammatory response follows (Hogg 1981), and the neutrophils and eosinophils release mediators that cause further oxidative injury (Castleman et al. 1980). Type II pneumocytes and cuboidal cells act as stem cells for repair (Keenan, Combs and McDowell 1982; Keenan, Wilson and McDowell 1983).
Other mechanisms of lung injury eventually involve the oxidative pathway of cellular damage, particularly after damage to the protective epithelial cell layer has occurred and an inflammatory response has been elicited. The most commonly described mechanisms are outlined in table 2.
Table 2. Mechanisms of lung injury by inhaled substances
|
Mechanism of injury |
Example compounds |
Damage that occurs |
|
Oxidation |
Ozone, nitrogen dioxide, sulphur dioxide, chlorine, oxides |
Patchy airway epithelial damage, with increased permeability and exposure of nerve fibre endings; loss of cilia from ciliated cells; necrosis of type I pneumocytes; free radical formation and subsequent protein binding and lipid peroxidation |
|
Acid formation |
Sulphur dioxide, chlorine, halides |
Gas dissolves in water to form acid that damages epithelial cells via oxidation; action mainly on upper airway |
|
Alkali formation |
Ammonia, calcium oxide, hydroxides |
Gas dissolves in water to form alkaline solution that may cause tissue liquefaction; predominant upper airway damage, lower airway in heavy exposures |
|
Protein binding |
Formaldehyde |
Reactions with amino acids lead to toxic intermediates with damage to the epithelial cell layer |
|
Afferent nerve stimulation |
Ammonia, formaldehyde |
Direct nerve ending stimulation provokes symptoms |
|
Antigenicity |
Platinum, acid anhydrides |
Low molecular weight molecules serve as haptens in sensitized persons |
|
Stimulation of host inflammatory response |
Copper and zinc oxides, lipoproteins |
Stimulation of cytokines and inflammatory mediators without apparent direct cellular damage |
|
Free radical formation |
Paraquat |
Promotion of formation or retardation of clearance of superoxide radicals, leading to lipid peroxidation and oxidative damage |
|
Delayed particle clearance |
Any prolonged inhalation of mineral dust |
Overwhelming of mucociliary escalators and alveolar macrophage systems with particles, leading to a non-specific inflammatory response |
Workers exposed to low levels of respiratory irritants may have subclinical symptoms traceable to mucous membrane irritation, such as watery eyes, sore throat, runny nose and cough. With significant exposure, the added feeling of shortness of breath will often prompt medical attention. It is important to secure a good medical history in order to determine the likely composition of the exposure, the quantity of exposure, and the period of time during which the exposure took place. Signs of laryngeal oedema, including hoarseness and stridor, should be sought, and the lungs should be examined for signs of lower airway or parenchymal involvement. Assessment of the airway and lung function, together with chest radiography, are important in short-term management. Laryngoscopy may be indicated to evaluate the airway.
If the airway is threatened, the patient should undergo intubation and supportive care. Patients with signs of laryngeal oedema should be observed for at least 12 hours to insure that the process is self-limited. Bronchospasm should be treated with b-agonists and, if refractory, intravenous corticosteroids. Irritated oral and ocular mucosa should be thoroughly irrigated. Patients with crackles on examination or chest radiograph abnormalities should be hospitalized for observation in view of the possibility of pneumonitis or pulmonary oedema. Such patients are at risk of bacterial superinfection; nevertheless, no benefit has been demonstrated by using prophylactic antibiotics.
The overwhelming majority of patients who survive the initial insult recover fully from irritant exposures. The chances for long-term sequelae are more likely with greater initial injury. The term reactive airway dysfunction syndrome (RADS) has been applied to the persistence of asthma-like symptoms following acute exposure to respiratory irritants (Brooks, Weiss and Bernstein 1985).
High-level exposures to alkalis and acids can cause upper and lower respiratory tract burns that lead to chronic disease. Ammonia is known to cause bronchiectasis (Kass et al. 1972); chlorine gas (which becomes HCl in the mucosa) is reported to cause obstructive lung disease (Donelly and Fitzgerald 1990; Das and Blanc 1993). Chronic low-level exposures to irritants may cause continued ocular and upper airway symptoms (Korn, Dockery and Speizer 1987), but deterioration of lung function has not been conclusively documented. Studies of the effects of chronic low-level irritants on airway function are hampered by a lack of long-term follow-up, confounding by cigarette smoking, the “healthy worker effect,” and the minimal, if any, actual clinical effect (Brooks and Kalica 1987).
After a patient recovers from the initial injury, regular follow-up by a physician is needed. Clearly, there should be an effort to investigate the workplace and evaluate respiratory precautions, ventilation and containment of the culprit irritants.
Toxic Chemicals
Chemicals toxic to the lung include most of the respiratory irritants given enough high exposure, but there are many chemicals that cause significant parenchymal lung injury despite possessing low to moderate irritant properties. These compounds work their effects by mechanisms reviewed in Table 3 and discussed above. Pulmonary toxins tend to be less water soluble than upper airway irritants. Examples of lung toxins and their sources of exposure are reviewed in table 3.
Table 3. Compounds capable of lung toxicity after low to moderate exposure
|
Compound |
Sources of exposure |
Toxicity |
|
Acrolein |
Plastics, textiles, pharmaceutical manufacturing, combustion products |
Diffuse airway and parenchymal injury |
|
Antimony trichloride; antimony |
Alloys, organic catalysts |
Pneumonitis, non-cardiogenic pulmonary oedema |
|
Cadmium |
Alloys with zinc and lead, electroplating, batteries, insecticides |
Tracheobronchitis, pulmonary oedema (often delayed onset over 24–48 hours), kidney damage: tubule proteinuria |
|
Chloropicrin |
Chemical manufacturing, fumigant components |
Upper and lower airway inflammation |
|
Chlorine |
Bleaching, formation of chlorinated compounds, household cleaners |
Upper and lower airway inflammation, pneumonitis and non-cardiogenic pulmonary oedema |
|
Hydrogen sulphide |
Natural gas wells, mines, manure |
Ocular, upper and lower airway irritation, delayed pulmonary oedema, asphyxiation from systemic tissue hypoxia |
|
Lithium hydride |
Alloys, ceramics, electronics, chemical catalysts |
Pneumonitis, non-cardiogenic pulmonary oedema |
|
Methyl isocyanate |
Pesticide synthesis |
Upper and lower respiratory tract irritation, pulmonary oedema |
|
Mercury |
Electrolysis, ore and amalgam extraction, electronics manufacture |
Ocular and respiratory tract inflammation, pneumonitis, CNS, kidney and systemic effects |
|
Nickel carbonyl |
Nickel refining, electroplating, chemical reagents |
Lower respiratory irritation, pneumonitis, delayed systemic toxic effects |
|
Nitrogen dioxide |
Silos after new grain storage, fertilizer making, arc welding; combustion products |
Ocular and upper airway inflammation, non-cardiogenic pulmonary oedema, delayed onset bronchiolitis |
|
Nitrogen mustards, sulphur |
Military agents, vesicants |
Ocular and respiratory tract inflammation, pneumonitis |
|
Paraquat |
Herbicides (ingested) |
Selective damage to type-2 pneumocytes leading to RADS, pulmonary fibrosis; renal failure, GI irritation |
|
Phosgene |
Pesticide and other chemical manufacture, arc welding, paint removal |
Upper airway inflammation and pneumonitis; delayed pulmonary oedema in low doses |
|
Zinc chloride |
Smoke grenades, artillery |
Upper and lower airway irritation, fever, delayed onset pneumonitis |
One group of inhalable toxins are termed asphyxiants. When present in high enough concentrations, the asphyxiants, carbon dioxide, methane and nitrogen, displace oxygen and in effect suffocate the victim. Hydrogen cyanide, carbon monoxide and hydrogen sulphide act by inhibiting cellular respiration despite adequate delivery of oxygen to the lung. Non-asphyxiant inhaled toxins damage target organs, causing a wide variety of health problems and mortality.
The medical management of inhaled lung toxins is similar to the management of respiratory irritants. These toxins often do not elicit their peak clinical effect for several hours after exposure; overnight monitoring may be indicated for compounds known to cause delayed onset pulmonary oedema. Since the therapy of systemic toxins is beyond the scope of this chapter, the reader is referred to discussions of the individual toxins elsewhere in this Encyclopaedia and in further texts on the subject (Goldfrank et al. 1990; Ellenhorn and Barceloux 1988).
Inhalation Fevers
Certain inhalation exposures occurring in a variety of different occupational settings may result in debilitating flu-like illnesses lasting a few hours. These are collectively referred to as inhalation fevers. Despite the severity of the symptoms, the toxicity seems to be self-limited in most cases, and there are few data to suggest long-term sequelae. Massive exposure to inciting compounds can cause a more severe reaction involving pneumonitis and pulmonary oedema; these uncommon cases are considered more complicated than simple inhalation fever.
The inhalation fevers have in common the feature of nonspecificity: the syndrome can be produced in nearly anyone, given adequate exposure to the inciting agent. Sensitization is not required, and no previous exposure is necessary. Some of the syndromes exhibit the phenomenon of tolerance; that is, with regular repeated exposure the symptoms do not occur. This effect is thought to be related to an increased activity of clearance mechanisms, but has not been adequately studied.
Organic Dust Toxic Syndrome
Organic dust toxic syndrome (ODTS) is a broad term denoting the self-limited flu-like symptoms that occur following heavy exposure to organic dusts. The syndrome encompasses a wide range of acute febrile illnesses that have names derived from the specific tasks that lead to dust exposure. Symptoms occur only after a massive exposure to organic dust, and most individuals so exposed will develop the syndrome.
Organic dust toxic syndrome has previously been called pulmonary mycotoxicosis, owing to its putative aetiology in the action of mould spores and actinomycetes. With some patients, one can culture species of Aspergillus, Penicillium, and mesophilic and thermophilic actinomycetes (Emmanuel, Marx and Ault 1975; Emmanuel, Marx and Ault 1989). More recently, bacterial endotoxins have been proposed to play at least as large a role. The syndrome has been provoked experimentally by inhalation of endotoxin derived from Enterobacter agglomerans, a major component of organic dust (Rylander, Bake and Fischer 1989). Endotoxin levels have been measured in the farm environment, with levels ranging from 0.01 to 100μg/m3. Many samples had a level greater than 0.2μg/m3, which is the level where clinical effects are known to occur (May, Stallones and Darrow 1989). There is speculation that cytokines, such as IL-1, may mediate the systemic effects, given what is already known about the release of IL-1 from alveolar macrophages in the presence of endotoxin (Richerson 1990). Allergic mechanisms are unlikely given the lack of need for sensitization and the requirement for high dust exposure.
Clinically, the patient will usually present symptoms 2 to 8 hours after exposure to (usually mouldy) grain, hay, cotton, flax, hemp or wood chips, or upon manipulation of pigs (Do Pico 1992). Often symptoms begin with eye and mucous membrane irritation with dry cough, progressing to fever, and malaise, chest tightness, myalgias and headache. The patient appears ill but otherwise normal upon physical examination. Leukocytosis frequently occurs, with levels as high as 25,000 white blood corpuscles (WBC)/mm3. The chest radiograph is almost always normal. Spirometry may reveal a modest obstructive defect. In cases where fibre optic bronchoscopy was performed and bronchial washings were obtained, an elevation of leukocytes was found in the lavage fluid. The percentage of neutrophils was significantly higher than normal (Emmanuel, Marx and Ault 1989; Lecours, Laviolette and Cormier 1986). Bronchoscopy 1 to 4 weeks after the event shows a persistently high cellularity, predominantly lymphocytes.
Depending on the nature of the exposure, the differential diagnosis may include toxic gas (such as nitrogen dioxide or ammonia) exposure, particularly if the episode occurred in a silo. Hypersensitivity pneumonitis should be considered, especially if there are significant chest radiograph or pulmonary function test abnormalities. The distinction between hypersensitivity pneumonitis (HP) and ODTS is important: HP will require strict exposure avoidance and has a worse prognosis, whereas ODTS has a benign and self-limited course. ODTS is also distinguished from HP because it occurs more frequently, requires higher levels of dust exposure, does not induce the release of serum precipitating antibodies, and (initially) does not give rise to the lymphocytic alveolitis that is characteristic of HP.
Cases are managed with antipyretics. A role for steroids has not been advocated given the self-limited nature of the illness. Patients should be educated about massive exposure avoidance. The long-term effect of repeated occurrences is thought to be negligible; however, this question has not been adequately studied.
Metal Fume Fever
Metal fume fever (MFF) is another self-limited, flu-like illness that develops after inhalation exposure, in this instance to metal fumes. The syndrome most commonly develops after zinc oxide inhalation, as occurs in brass foundries, and in smelting or welding galvanized metal. Oxides of copper and iron also cause MFF, and vapours of aluminium, arsenic, cadmium, mercury, cobalt, chromium, silver, manganese, selenium and tin have been occasionally implicated (Rose 1992). Workers develop tachyphalaxis; that is, symptoms appear only when the exposure occurs after several days without exposure, not when there are regular repeated exposures. An eight-hour TLV of 5 mg/m3 for zinc oxide has been established by the US Occupational Safety and Health Administration (OSHA), but symptoms have been elicited experimentally after a two-hour exposure at this concentration (Gordon et al. 1992).
The pathogenesis of MFF remains unclear. The reproducible onset of symptoms regardless of the individual exposed argues against a specific immune or allergic sensitization. The lack of symptoms associated with histamine release (flushing, itching, wheezing, hives) also militates against the likelihood of an allergic mechanism. Paul Blanc and co-workers have developed a model implicating cytokine release (Blanc et al. 1991; Blanc et al.1993). They measured the levels of tumour necrosis factor (TNF), and of the interleukins IL-1, IL-4, IL-6 and IL-8 in the fluid lavaged from the lungs of 23 volunteers experimentally exposed to zinc oxide fumes (Blanc et al. 1993). The volunteers developed elevated levels of TNF in their bronchoalveolar lavage (BAL) fluid 3 hours after exposure. Twenty hours later, high BAL fluid levels of IL-8 (a potent neutrophil attractant) and an impressive neutrophilic alveolitis were observed. TNF, a cytokine capable of causing fever and stimulating immune cells, has been shown to be released from monocytes in culture that are exposed to zinc (Scuderi 1990). Accordingly, the presence of increased TNF in the lung accounts for the onset of symptoms observed in MFF. TNF is known to stimulate the release of both IL-6 and IL-8, in a time period that correlated with the peaks of the cytokines in these volunteers’ BAL fluid. The recruitment of these cytokines may account for the ensuing neutrophil alveolitis and flu-like symptoms that characterize MFF. Why the alveolitis resolves so quickly remains a mystery.
Symptoms begin 3 to 10 hours after exposure. Initially, there may be a sweet metallic taste in the mouth, accompanied by a worsening dry cough and shortness of breath. Fever and shaking chills often develop, and the worker feels ill. The physical examination is otherwise unremarkable. Laboratory evaluation shows a leukocytosis and a normal chest radiograph. Pulmonary function studies may show a slightly reduced FEF25-75 and DLCO levels (Nemery 1990; Rose 1992).
With a good history the diagnosis is readily established and the worker can be treated symptomatically with antipyretics. Symptoms and clinical abnormalities resolve within 24 to 48 hours. Otherwise, bacterial and viral aetiologies of the symptoms must be considered. In cases of extreme exposure, or exposures involving contamination by toxins such as zinc chloride, cadmium or mercury, MFF may be a harbinger of a clinical chemical pneumonitis that will evolve over the next 2 days (Blount 1990). Such cases can exhibit diffuse infiltrates on a chest radiograph and signs of pulmonary oedema and respiratory failure. While this possibility should be considered in the initial evaluation of an exposed patient, such a fulminant course is unusual and not characteristic of uncomplicated MFF.
MFF does not require a specific sensitivity of the individual for the metal fumes; rather, it indicates inadequate environmental control. The exposure problem should be addressed to prevent recurrent symptoms. Although the syndrome is considered benign, the long-term effects of repeated bouts of MFF have not been adequately investigated.
Polymer Fume Fever
Polymer fume fever is a self-limited febrile illness similar to MFF, but caused by inhaled pyrolysis products of fluoropolymers, including polytetrafluoroethane (PTFE; trade names Teflon, Fluon, Halon). PTFE is widely used for its lubricant, thermal stability and electrical insulative properties. It is harmless unless heated above 30°C, when it starts to release degradation products (Shusterman 1993). This situation occurs when welding materials coated with PTFE, heating PTFE with a tool edge during high speed machining, operating moulding or extruding machines (Rose 1992) and rarely during endotracheal laser surgery (Rom 1992a).
A common cause of polymer fume fever was elicited after a period of classic public health detective work in the early 1970s (Wegman and Peters 1974; Kuntz and McCord 1974). Textile workers were developing self-limited febrile illnesses with exposures to formaldehyde, ammonia and nylon fibre; they did not have exposure to fluoropolymer fumes but handled crushed polymer. After finding that exposure levels of the other possible aetiological agents were within acceptable limits, the fluoropolymer work was examined more closely. As it turned out, only cigarette smokers working with the fluoropolymer were symptomatic. It was hypothesized that the cigarettes were being contaminated with fluoropolymer on the worker’s hands, then the product was combusted on the cigarette when it was smoked, exposing the worker to toxic fumes. After banning cigarette smoking in the workplace and setting strict handwashing rules, no further illnesses were reported (Wegman and Peters 1974). Since then, this phenomenon has been reported after working with waterproofing compounds, mould-release compounds (Albrecht and Bryant 1987) and after using certain kinds of ski wax (Strom and Alexandersen 1990).
The pathogenesis of polymer fume fever is not known. It is thought to be similar to the other inhalation fevers owing to its similar presentation and apparently non-specific immune response. There have been no human experimental studies; however, rats and birds both develop severe alveolar epithelial damage on exposure to PTFE pyrolysis products (Wells, Slocombe and Trapp 1982; Blandford et al. 1975). Accurate measurement of pulmonary function or BAL fluid changes has not been done.
Symptoms appear several hours after exposure, and a tolerance or tachyphalaxis effect is not there as seen in MFF. Weakness and myalgias are followed by fever and chills. Often there is chest tightness and cough. Physical examination is usually otherwise normal. Leukocytosis is often seen, and the chest radiograph is usually normal. Symptoms resolve spontaneously in 12 to 48 hours. There have been a few cases of persons developing pulmonary oedema after exposure; in general, PTFE fumes are thought to be more toxic than zinc or copper fumes in causing MFF (Shusterman 1993; Brubaker 1977). Chronic airways dysfunction has been reported in persons who have had multiple episodes of polymer fume fever (Williams, Atkinson and Patchefsky 1974).
The diagnosis of polymer fume fever requires a careful history with high clinical suspicion. After ascertaining the source of the PTFE pyrolysis products, efforts must be made to prevent further exposure. Mandatory handwashing rules and the elimination of smoking in the workplace has effectively eliminated cases related to contaminated cigarettes. Workers who have had multiple episodes of polymer fume fever or associated pulmonary oedema should have long-term medical follow-up.
Occupational Asthma
Asthma is a respiratory disease characterized by airway obstruction that is partially or completely reversible, either spontaneously or with treatment; airway inflammation; and increased airway responsiveness to a variety of stimuli (NAEP 1991). Occupational asthma (OA) is asthma that is caused by environmental exposures in the workplace. Several hundred agents have been reported to cause OA. Pre-existing asthma or airway hyper-responsiveness, with symptoms worsened by work exposure to irritants or physical stimuli, is usually classified separately as work-aggravated asthma (WAA). There is general agreement that OA has become the most prevalent occupational lung disease in developed countries, although estimates of actual prevalence and incidence are quite variable. It is clear, however, that in many countries asthma of occupational aetiology causes a largely unrecognized burden of disease and disability with high economic and non-economic costs. Much of this public health and economic burden is potentially preventable by identifying and controlling or eliminating the workplace exposures causing the asthma. This article will summarize current approaches to recognition, management and prevention of OA. Several recent publications discuss these issues in more detail (Chan-Yeung 1995; Bernstein et al. 1993).
Magnitude of the Problem
Prevalences of asthma in adults generally range from 3 to 5%, depending on the definition of asthma and geographic variations, and may be considerably higher in some low-income urban populations. The proportion of adult asthma cases in the general population that is related to the work environment is reported to range from 2 to 23%, with recent estimates tending towards the higher end of the range. Prevalences of asthma and OA have been estimated in small cohort and cross-sectional studies of high-risk occupational groups. In a review of 22 selected studies of workplaces with exposures to specific substances, prevalences of asthma or OA, defined in various ways, ranged from 3 to 54%, with 12 studies reporting prevalences over 15% (Becklake, in Bernstein et al. 1993). The wide range reflects real variation in actual prevalence (due to different types and levels of exposure). It also reflects differences in diagnostic criteria, and variation in the strength of the biases, such as “survivor bias” which may result from exclusion of workers who developed OA and left the workplace before the study was conducted. Population estimates of incidence range from 14 per million employed adults per year in the United States to 140 per million employed adults per year in Finland (Meredith and Nordman 1996). Ascertainment of cases was more complete and methods of diagnosis were generally more rigorous in Finland. The evidence from these different sources is consistent in its implication that OA is often under-diagnosed and/or under-reported and is a public health problem of greater magnitude than generally recognized.
Causes of Occupational Asthma
Over 200 agents (specific substances, occupations or industrial processes) have been reported to cause OA, based on epidemiological and/or clinical evidence. In OA, airway inflammation and bronchoconstriction can be caused by immunological response to sensitizing agents, by direct irritant effects, or by other non-immunological mechanisms. Some agents (e.g., organophosphate insecticides) may also cause bronchoconstriction by direct pharmacological action. Most of the reported agents are thought to induce a sensitization response. Respiratory irritants often worsen symptoms in workers with pre-existing asthma (i.e., WAA) and, at high exposure levels, can cause new onset of asthma (termed reactive airways dysfunction syndrome (RADS) or irritant-induced asthma) (Brooks, Weiss and Bernstein 1985; Alberts and Do Pico 1996).
OA may occur with or without a latency period. Latency period refers to the time between initial exposure and development of symptoms, and is highly variable. It is often less than 2 years, but in around 20% of cases is 10 years or longer. OA with latency is generally caused by sensitization to one or more agents. RADS is an example of OA without latency.
High molecular weight sensitizing agents (5,000 daltons (Da) or greater) often act by an IgE-dependent mechanism. Low molecular weight sensitizing agents (less than 5,000 Da), which include highly reactive chemicals like isocyanates, may act by IgE-independent mechanisms or may act as haptens, combining with body proteins. Once a worker becomes sensitized to an agent, re-exposure (frequently at levels far below the level that caused sensitization) results in an inflammatory response in the airways, often accompanied by increases in airflow limitation and non-specific bronchial responsiveness (NBR).
In epidemiological studies of OA, workplace exposures are consistently the strongest determinants of asthma prevalence, and the risk of developing OA with latency tends to increase with estimated intensity of exposure. Atopy is an important and smoking a somewhat less consistent determinant of asthma occurrence in studies of agents that act through an IgE-dependent mechanism. Neither atopy nor smoking appears to be an important determinant of asthma in studies of agents acting through IgE-independent mechanisms.
Clinical Presentation
The symptom spectrum of OA is similar to non-occupational asthma: wheeze, cough, chest tightness and shortness of breath. Patients sometimes present cough-variant or nocturnal asthma. OA can be severe and disabling, and deaths have been reported. Onset of OA occurs due to a specific job environment, so identifying exposures that occurred at the time of onset of asthmatic symptoms is key to an accurate diagnosis. In WAA, workplace exposures cause a significant increase in frequency and/or severity of symptoms of pre-existing asthma.
Several features of the clinical history may suggest occupational aetiology (Chan-Yeung 1995). Symptoms frequently worsen at work or at night after work, improve on days off, and recur on return to work. Symptoms may worsen progressively towards the end of the workweek. The patient may note specific activities or agents in the workplace that reproducibly trigger symptoms. Work-related eye irritation or rhinitis may be associated with asthmatic symptoms. These typical symptom patterns may be present only in the initial stages of OA. Partial or complete resolution on weekends or vacations is common early in the course of OA, but with repeated exposures, the time required for recovery may increase to one or two weeks, or recovery may cease to occur. The majority of patients with OA whose exposures are terminated continue to have symptomatic asthma even years after cessation of exposure, with permanent impairment and disability. Continuing exposure is associated with further worsening of asthma. Brief duration and mild severity of symptoms at the time of cessation of exposure are good prognostic factors and decrease the likelihood of permanent asthma.
Several characteristic temporal patterns of symptoms have been reported for OA. Early asthmatic reactions typically occur shortly (less than one hour) after beginning work or the specific work exposure causing the asthma. Late asthmatic reactions begin 4 to 6 hours after exposure begins, and can last 24 to 48 hours. Combinations of these patterns occur as dual asthmatic reactions with spontaneous resolution of symptoms separating an early and late reaction, or as continuous asthmatic reactions with no resolution of symptoms between phases. With exceptions, early reactions tend to be IgE mediated, and late reactions tend to be IgE independent.
Increased NBR, generally measured by methacholine or histamine challenge, is considered a cardinal feature of occupational asthma. The time course and degree of NBR may be useful in diagnosis and monitoring. NBR may decrease within several weeks after cessation of exposure, although abnormal NBR commonly persists for months or years after exposures are terminated. In individuals with irritant-induced occupational asthma, NBR is not expected to vary with exposure and/or symptoms.
Recognition and Diagnosis
Accurate diagnosis of OA is important, given the substantial negative consequences of either under- or over-diagnosis. In workers with OA or at risk of developing OA, timely recognition, identification and control of the occupational exposures causing the asthma improve the chances of prevention or complete recovery. This primary prevention can greatly reduce the high financial and human costs of chronic, disabling asthma. Conversely, since a diagnosis of OA may obligate a complete change of occupation, or costly interventions in the workplace, accurately distinguishing OA from asthma that is not occupational can prevent unnecessary social and financial costs to both employers and workers.
Several case definitions of OA have been proposed, appropriate in different circumstances. Definitions found valuable for worker screening or surveillance (Hoffman et al. 1990) may not be entirely applicable for clinical purposes or compensation. A consensus of researchers has defined OA as “a disease characterized by variable airflow limitation and/or airway hyper-responsiveness due to causes and conditions attributable to a particular occupational environment and not to stimuli encountered outside the workplace” (Bernstein et al. 1993). This definition has been operationalized as a medical case definition, summarized in table 1 (Chan-Yeung 1995).
Table 1. ACCP medical case definition of occupational asthma
Criteria for diagnosis of occupational asthma1 (requires all 4, A-D):
(A) Physician diagnosis of asthma and/or physiological evidence of airways hyper-responsiveness
(B) Occupational exposure preceded onset of asthmatic symptoms1
(C) Association between symptoms of asthma and work
(D) Exposure and/or physiological evidence of relation of asthma to workplace environment (Diagnosis of OA requires one or more of D2-D5, likely OA requires only D1)
(1) Workplace exposure to agent reported to give rise to OA
(2) Work-related changes in FEV1 and/or PEF
(3) Work-related changes in serial testing for non-specific bronchial responsiveness (e.g., Methacholine Challenge Test)
(4) Positive specific bronchial challenge test
(5) Onset of asthma with a clear association with a symptomatic exposure to an inhaled irritant in the workplace (generally RADS)
Criteria for diagnosis of RADS (should meet all 7):
(1) Documented absence of preexisting asthma-like complaints
(2) Onset of symptoms after a single exposure incident or accident
(3) Exposure to a gas, smoke, fume, vapour or dust with irritant properties present in high concentration
(4) Onset of symptoms within 24 hours after exposure with persistence of symptoms for at least 3 months
(5) Symptoms consistent with asthma: cough, wheeze, dyspnoea
(6) Presence of airflow obstruction on pulmonary function tests and/or presence of non-specific bronchial hyper-responsiveness (testing should be done shortly after exposure)
(7) Other pulmonary diseases ruled out
Criteria for diagnosis of work-aggravated asthma (WAA):
(1) Meets criteria A and C of ACCP Medical Case Definition of OA
(2) Pre-existing asthma or history of asthmatic symptoms, (with active symptoms during the year prior to start of employment or exposure of interest)
(3) Clear increase in symptoms or medication requirement, or documentation of work-related changes in PEFR or FEV1 after start of employment or exposure of interest
1 A case definition requiring A, C and any one of D1 to D5 may be useful in surveillance for OA, WAA and RADS.
Source: Chan-Yeung 1995.
Thorough clinical evaluation of OA can be time consuming, costly and difficult. It may require diagnostic trials of removal from and return to work, and often requires the patient to reliably chart serial peak expiratory flow (PEF) measurements. Some components of the clinical evaluation (e.g., specific bronchial challenge or serial quantitative testing for NBR) may not be readily available to many physicians. Other components may simply not be achievable (e.g., patient no longer working, diagnostic resources not available, inadequate serial PEF measurements). Diagnostic accuracy is likely to increase with the thoroughness of the clinical evaluation. In each individual patient, decisions on the extent of medical evaluation will need to balance costs of the evaluation with the clinical, social, financial and public health consequences of incorrectly diagnosing or ruling out OA.
In consideration of these difficulties, a stepped approach to diagnosis of OA is outlined in table 2. This is intended as a general guide to facilitate accurate, practical and efficient diagnostic evaluation, recognizing that some of the suggested procedures may not be available in some settings. Diagnosis of OA involves establishing both the diagnosis of asthma and the relation between asthma and workplace exposures. After each step, for each patient, the physician will need to determine whether the level of diagnostic certainty achieved is adequate to support the necessary decisions, or whether evaluation should continue to the next step. If facilities and resources are available, the time and cost of continuing the clinical evaluation are usually justified by the importance of making an accurate determination of the relationship of asthma to work. Highlights of diagnostic procedures for OA will be summarized; details can be found in several of the references (Chan-Yeung 1995; Bernstein et al. 1993). Consultation with a physician experienced in OA may be considered, since the diagnostic process may be difficult.
Table 2. Steps in diagnostic evaluation of asthma in the workplace
Step 1 Thorough medical and occupational history and directed physical examination.
Step 2 Physiologic evaluation for reversible airway obstruction and/or non specific bronchial hyper-responsiveness.
Step 3 Immunologic assessment, if appropriate.
Assess Work Status:
Currently working: Proceed to Step 4 first.
Not currently working, diagnostic trial of return to work feasible: Step 5 first, then Step 4.
Not currently working, diagnostic trial of return to work not feasible: Step 6.
Step 4 Clinical evaluation of asthma at work or diagnostic trial of return to work.
Step 5 Clinical evaluation of asthma away from work or diagnostic trial of removal from work.
Step 6 Workplace challenge or specific bronchial challenge testing. If available for suspected causal exposures, this step may be performed prior to Step 4 for any patient.
This is intended as a general guide to facilitate practical and efficient diagnostic evaluation. It is recommended that physicians who diagnose and manage OA refer to current clinical literature as well.
RADS, when caused by an occupational exposure, is usually considered a subclass of OA. It is diagnosed clinically, using the criteria in Table 6. Patients who have experienced significant respiratory injury due to high-level irritant inhalations should be evaluated for persistent symptoms and presence of airflow obstruction shortly after the event. If the clinical history is compatible with RADS, further evaluation should include quantitative testing for NBR, if not contra-indicated.
WAA may be common, and may cause a substantial preventable burden of disability, but little has been published on diagnosis, management or prognosis. As summarized in Table 6, WAA is recognized when asthmatic symptoms preceded the suspected causal exposure but are clearly aggravated by the work environment. Worsening at work can be documented either by physiological evidence or through evaluation of medical records and medication use. It is a clinical judgement whether patients with a history of asthma in remission, who have recurrence of asthmatic symptoms that otherwise meet the criteria for OA, are diagnosed with OA or WAA. One year has been proposed as a sufficiently long asymptomatic period that the onset of symptoms is likely to represent a new process caused by the workplace exposure, although no consensus yet exists.
Step 1: Thorough medical and occupational history anddirected physical examination
Initial suspicion of possible OA in appropriate clinical and workplace situations is key, given the importance of early diagnosis and intervention in improving prognosis. The diagnosis of OA or WAA should be considered in all asthmatic patients in whom symptoms developed as a working adult (especially recent onset), or in whom the severity of asthma has substantially increased. OA should also be considered in any other individuals who have asthma-like symptoms and work in occupations in which they are exposed to asthma-causing agents or who are concerned that their symptoms are work-related.
Patients with possible OA should be asked to provide a thorough medical and occupational/environmental history, with careful documentation of the nature and date of onset of symptoms and diagnosis of asthma, and any potentially causal exposures at that time. Compatibility of the medical history with the clinical presentation of OA described above should be evaluated, especially the temporal pattern of symptoms in relation to work schedule and changes in work exposures. Patterns and changes in patterns of use of asthma medications, and the minimum period of time away from work required for improvement in symptoms should be noted. Prior respiratory diseases, allergies/atopy, smoking and other toxic exposures, and a family history of allergy are pertinent.
Occupational and other environmental exposures to potential asthma-causing agents or processes should be thoroughly explored, with objective documentation of exposures if possible. Suspected exposures should be compared with a comprehensive list of agents reported to cause OA (Harber, Schenker and Balmes 1996; Chan-Yeung and Malo 1994; Bernstein et al. 1993; Rom 1992b), although inability to identify specific agents is not uncommon and induction of asthma by agents not previously described is possible as well. Some illustrative examples are shown in table 3. Occupational history should include details of current and relevant past employment with dates, job titles, tasks and exposures, especially current job and job held at time of onset of symptoms. Other environmental history should include a review of exposures in the home or community that could cause asthma. It is helpful to begin the exposure history in an open-ended way, asking about broad categories of airborne agents: dusts (especially organic dusts of animal, plant or microbial origin), chemicals, pharmaceuticals and irritating or visible gases or fumes. The patient may identify specific agents, work processes or generic categories of agents that have triggered symptoms. Asking the patient to describe step by step the activities and exposures involved in the most recent symptomatic workday can provide useful clues. Materials used by co-workers, or those released in high concentration from a spill or other source, may be relevant. Further information can often be obtained on product name, ingredients and manufacturer name, address and phone number. Specific agents can be identified by calling the manufacturer or through a variety of other sources including textbooks, CD ROM databases, or Poison Control Centers. Since OA is frequently caused by low levels of airborne allergens, workplace industrial hygiene inspections which qualitatively evaluate exposures and control measures are often more helpful than quantitative measurement of air contaminants.
Table 3. Sensitizing agents that can cause occupational asthma
|
Classification |
Sub-groups |
Examples of substances |
Examples of jobs and industries |
|
High-molecular-weight protein antigens |
Animal-derived substances Plant-derived substances |
Laboratory animals, crab/seafood, mites, insects Flour and grain dusts, natural rubber latex gloves, bacterial enzymes, castor bean dust, vegetable gums |
Animal handlers, farming and food processing Bakeries, health care workers, detergent making, food processing |
|
Low-molecular-weight/chemical |
Plasticizers, 2-part paints, adhesives, foams Metals Wood dusts Pharmaceuticals, drugs |
Isocyanates, acid anhydrides, amines Platinum salts, cobalt Cedar (plicatic acid), oak Psyllium, antibiotics |
Auto spray painting, varnishing, woodworking Platinum refineries, metal grinding Sawmill work, carpentry Pharmaceutical manufacturing and packaging |
|
Other chemicals |
Chloramine T, polyvinyl chloride fumes, organophosphate insecticides |
Janitorial work, meat packing |
The clinical history appears to be better for excluding rather than for confirming the diagnosis of OA, and an open-ended history taken by a physician is better than a closed questionnaire. One study compared the results of an open-ended clinical history taken by trained OA specialists with a “gold standard” of specific bronchial challenge testing in 162 patients referred for evaluation of possible OA. The investigators reported that the sensitivity of a clinical history suggestive of OA was 87%, specificity 55%, predictive value positive 63% and predictive value negative 83%. In this group of referred patients, prevalence of asthma and OA were 80% and 46%, respectively (Malo et al. 1991). In other groups of referred patients, predictive values positive of a closed questionnaire ranged from 8 to 52% for a variety of workplace exposures (Bernstein et al. 1993). The applicability of these results to other settings needs to be assessed by the physician.
Physical examination is sometimes helpful, and findings relevant to asthma (e.g., wheezing, nasal polyps, eczematous dermatitis), respiratory irritation or allergy (e.g., rhinitis, conjunctivitis) or other potential causes of symptoms should be noted.
Step 2: Physiological evaluation for reversible airway obstruction and/or non-specific bronchial hyper-responsiveness
If sufficient physiological evidence supporting the diagnosis of asthma (NAEP 1991) is already in the medical record, Step 2 can be skipped. If not, technician-coached spirometry should be performed, preferably post-workshift on a day when the patient is experiencing asthmatic symptoms. If spirometry reveals airway obstruction which reverses with a bronchodilator, this confirms the diagnosis of asthma. In patients without clear evidence of airflow limitation on spirometry, quantitative testing for NBR using methacholine or histamine should be done, the same day if possible. Quantitative testing for NBR in this situation is a key procedure for two reasons. First, it can often identify patients with mild or early stage OA who have the greatest potential for cure but who would be missed if testing stopped with normal spirometry. Second, if NBR is normal in a worker who has ongoing exposure in the workplace environment associated with the symptoms, OA can generally be ruled out without further testing. If abnormal, evaluation can proceed to Step 3 or 4, and the degree of NBR may be useful in monitoring the patient for improvement after diagnostic trial of removal from the suspected causal exposure (Step 5). If spirometry reveals significant airflow limitation that does not improve after inhaled bronchodilator, a re-evaluation after more prolonged trial of therapy, including corticosteroids, should be considered (ATS 1995; NAEP 1991).
Step 3: Immunological assessment, if appropriate
Skin or serological (e.g., RAST) testing can demonstrate immunological sensitization to a specific workplace agent. These immunological tests have been used to confirm the work-relatedness of asthma, and, in some cases, eliminate the need for specific inhalation challenge tests. For example, among psyllium-exposed patients with a clinical history compatible with OA, documented asthma or airway hyper-responsiveness, and evidence of immunological sensitization to psyllium, approximately 80% had OA confirmed on subsequent specific bronchial challenge testing (Malo et al. 1990). In most cases, diagnostic significance of negative immunological tests is less clear. The diagnostic sensitivity of the immunological tests depends critically on whether all the likely causal antigens in the workplace or hapten-protein complexes have been included in the testing. Although the implication of sensitization for an asymptomatic worker is not well defined, analysis of grouped results can be useful in evaluating environmental controls. The utility of immunological evaluation is greatest for agents for which there are standardized in vitro tests or skin-prick reagents, such as platinum salts and detergent enzymes. Unfortunately, most occupational allergens of interest are not currently available commercially. The use of non-commercial solutions in skin-prick testing has on occasions been associated with severe reactions, including anaphylaxis, and thus caution is necessary.
If results of Steps 1 and 2 are compatible with OA, further evaluation should be pursued if possible. The order and extent of further evaluation depends on availability of diagnostic resources, work status of the patient and feasibility of diagnostic trials of removal from and return to work as indicated in Table 7. If further evaluation is not possible, a diagnosis must be based on the information available at this point.
Step 4: Clinical evaluation of asthma at work, or diagnostic trial of return to work
Often the most readily available physiological test of airway obstruction is spirometry. To improve reproducibility, spirometry should be coached by a trained technician. Unfortunately, single-day cross-shift spirometry, performed before and after the workshift, is neither sensitive nor specific in determining work-associated airway obstruction. It is probable that if multiple spirometries are performed each day during and after several workdays, the diagnostic accuracy may be improved, but this has not yet been adequately evaluated.
Due to difficulties with cross-shift spirometry, serial PEF measurement has become an important diagnostic technique for OA. Using an inexpensive portable meter, PEF measurements are recorded every two hours, during waking hours. To improve sensitivity, measurements must be done during a period when the worker is exposed to the suspected causal agents at work and is experiencing a work-related pattern of symptoms. Three repetitions are performed at each time, and measurements are made every day at work and away from work. The measurements should be continued for at least 16 consecutive days (e.g., two five-day work weeks and 3 weekends off) if the patient can safely tolerate continuing to work. PEF measurements are recorded in a diary along with notation of work hours, symptoms, use of bronchodilator medications, and significant exposures. To facilitate interpretation, the diary results should then be plotted graphically. Certain patterns suggest OA, but none are pathognomonic, and interpretation by an experienced reader is often helpful. Advantages of serial PEF testing are low cost and reasonable correlation with results of bronchial challenge testing. Disadvantages include the significant degree of patient cooperation required, inability to definitely confirm that data are accurate, lack of standardized method of interpretation, and the need for some patients to take 1 or 2 consecutive weeks off work to show significant improvement. Portable electronic recording spirometers designed for patient self monitoring, when available, can address some of the disadvantages of serial PEF.
Asthma medications tend to reduce the effect of work exposures on measures of airflow. However, it is not advisable to discontinue medications during airflow monitoring at work. Rather, the patient should be maintained on a constant minimal safe dosage of anti-inflammatory medications throughout the entire diagnostic process, with close monitoring of symptoms and airflow, and the use of short-acting bronchodilators to control symptoms should be noted in the diary.
The failure to observe work-related changes in PEF while a patient is working routine hours does not exclude the diagnosis of OA, since many patients will require more than a two-day weekend to show significant improvement in PEF. In this case, a diagnostic trial of extended removal from work (Step 5) should be considered. If the patient has not yet had quantitative testing for NBR, and does not have a medical contra-indication, it should be done at this time, immediately after at least two weeks of workplace exposure.
Step 5: Clinical evaluation of asthma away from work or diagnostic trial of extended removal from work
This step consists of completion of the serial 2-hourly PEF daily diary for at least 9 consecutive days away from work (e.g., 5 days off work plus weekends before and after). If this record, compared with the serial PEF diary at work, is not sufficient for diagnosing OA, it should be continued for a second consecutive week away from work. After 2 or more weeks away from work, quantitative testing for NBR can be performed and compared to NBR while at work. If serial PEF has not yet been done during at least two weeks at work, then a diagnostic trial of return to work (see Step 4) may be performed, after detailed counselling, and in close contact with the treating physician. Step 5 is often critically important in confirming or excluding the diagnosis of OA, although it may also be the most difficult and expensive step. If an extended removal from work is attempted, it is best to maximize the diagnostic yield and efficiency by including PEF, FEV1, and NBR tests in one comprehensive evaluation. Weekly physician visits for counselling and to review the PEF chart can help to assure complete and accurate results. If, after monitoring the patient for at least two weeks at work and two weeks away from it, the diagnostic evidence is not yet sufficient, Step 6 should be considered next, if available and feasible.
Step 6: Specific bronchial challenge or workplace challenge testing
Specific bronchial challenge testing using an exposure chamber and standardized exposure levels has been labelled the “gold standard” for diagnosis of OA. Advantages include definitive confirmation of OA with ability to identify asthmatic response to sub-irritant levels of specific sensitizing agents, which can then be scrupulously avoided. Of all the diagnostic methods, it is the only one that can reliably distinguish sensitizer-induced asthma from provocation by irritants. Several problems with this approach have included inherent costliness of the procedure, general requirement of close observation or hospitalization for several days, and availability in only very few specialized centres. False negatives may occur if standardized methodology is not available for all suspected agents, if the wrong agents are suspected, or if too long a time has elapsed between last exposure and testing. False positives may result if irritant levels of exposure are inadvertently obtained. For these reasons, specific bronchial challenge testing for OA remains a research procedure in most localities.
Workplace challenge testing involves serial technician-coached spirometry in the workplace, performed at frequent (e.g., hourly) intervals before and during the course of a workday exposure to the suspected causal agents or processes. It may be more sensitive than specific bronchial challenge testing because it involves “real life” exposures, but since airway obstruction may be triggered by irritants as well as sensitizing agents, positive tests do not necessarily indicate sensitization. It also requires cooperation of the employer and much technician time with a mobile spirometer. Both of these procedures carry some risk of precipitating a severe asthmatic attack, and should therefore be done under close supervision of specialists experienced with the procedures.
Treatment and Prevention
Management of OA includes medical and preventive interventions for individual patients, as well as public health measures in workplaces identified as high risk for OA. Medical management is similar to that for non-occupational asthma and is well reviewed elsewhere (NAEP 1991). Medical management alone is rarely adequate to optimally control symptoms, and preventive intervention by control or cessation of exposure is an integral part of the treatment. This process begins with accurate diagnosis and identification of causative exposures and conditions. In sensitizer-induced OA, reducing exposure to the sensitizer does not usually result in complete resolution of symptoms. Severe asthmatic episodes or progressive worsening of the disease may be caused by exposures to very low concentrations of the agent and complete and permanent cessation of exposure is recommended. Timely referral for vocational rehabilitation and job retraining may be a necessary component of treatment for some patients. If complete cessation of exposure is impossible, substantial reduction of exposure accompanied by close medical monitoring and management may be an option, although such reduction in exposure is not always feasible and the long-term safety of this approach has not been tested. As an example, it would be difficult to justify the toxicity of long-term treatment with systemic corticosteroids in order to allow the patient to continue in the same employment. For asthma induced and/or triggered by irritants, dose response may be more predictable, and lowering of irritant exposure levels, accompanied by close medical monitoring, may be less risky and more likely to be effective than for sensitizer-induced OA. If the patient continues to work under modified conditions, medical follow-up should include frequent physician visits with review of the PEF diary, well-planned access to emergency services, and serial spirometry and/or methacholine challenge testing, as appropriate.
Once a particular workplace is suspected to be high risk, due either to occurrence of a sentinel case of OA or use of known asthma-causing agents, public health methods can be very useful. Early recognition and effective treatment and prevention of disability of workers with existing OA, and prevention of new cases, are clear priorities. Identification of specific causal agent(s) and work processes is important. One practical initial approach is a workplace questionnaire survey, evaluating criteria A, B, C, and D1 or D5 in the case definition of OA. This approach can identify individuals for whom further clinical evaluation might be indicated and help identify possible causal agents or circumstances. Evaluation of group results can help decide whether further workplace investigation or intervention is indicated and, if so, provide valuable guidance in targeting future prevention efforts in the most effective and efficient manner. A questionnaire survey is not adequate, however, to establish individual medical diagnoses, since predictive positive values of questionnaires for OA are not high enough. If a greater level of diagnostic certainty is needed, medical screening utilizing diagnostic procedures such as spirometry, quantitative testing for NBR, serial PEF recording, and immunological testing can be considered as well. In known problem workplaces, ongoing surveillance and screening programmes may be helpful. However, differential exclusion of asymptomatic workers with history of atopy or other potential susceptibility factors from workplaces believed to be high risk would result in removal of large numbers of workers to prevent relatively few cases of OA, and is not supported by the current literature.
Control or elimination of causal exposures and avoidance and proper management of spills or episodes of high-level exposures can lead to effective primary prevention of sensitization and OA in co-workers of the sentinel case. The usual exposure control hierarchy of substitution, engineering and administrative controls, and personal protective equipment, as well as education of workers and managers, should be implemented as appropriate. Proactive employers will initiate or participate in some or all of these approaches, but in the event that inadequate preventive action is taken and workers remain at high risk, governmental enforcement agencies may be helpful.
Impairment and Disability
Medical impairment is a functional abnormality resulting from a medical condition. Disability refers to the total effect of the medical impairment on the patient’s life, and is influenced by many non-medical factors such as age and socio-economic status (ATS 1995).
Assessment of medical impairment is done by the physician and may include a calculated impairment index, as well as other clinical considerations. The impairment index is based on (1) degree of airflow limitation after bronchodilator, (2) either degree of reversibility of airflow limitation with bronchodilator or degree of airway hyper-responsiveness on quantitative testing for NBR, and (3) minimum medication required to control asthma. The other major component of the assessment of medical impairment is the physician’s medical judgement of the ability of the patient to work in the workplace environment causing the asthma. For example, a patient with sensitizer-induced OA may have a medical impairment which is highly specific to the agent to which he or she has become sensitized. The worker who experiences symptoms only when exposed to this agent may be able to work in other jobs, but permanently unable to work in the specific job for which she or he has the most training and experience.
Assessment of disability due to asthma (including OA) requires consideration of medical impairment as well as other non-medical factors affecting ability to work and function in everyday life. Disability assessment is initially made by the physician, who should identify all the factors affecting the impact of the impairment on the patient’s life. Many factors such as occupation, educational level, possession of other marketable skills, economic conditions and other social factors may lead to varying levels of disability in individuals with the same level of medical impairment. This information can then be used by administrators to determine disability for purposes of compensation.
Impairment and disability may be classified as temporary or permanent, depending on the likelihood of significant improvement, and whether effective exposure controls are successfully implemented in the workplace. For example, an individual with sensitizer-induced OA is generally considered permanently, totally impaired for any job involving exposure to the causal agent. If the symptoms resolve partially or completely after cessation of exposure, these individuals may be classified with less or no impairment for other jobs. Often this is considered permanent partial impairment/disability, but terminology may vary. An individual with asthma which is triggered in a dose-dependent fashion by irritants in the workplace would be considered to have temporary impairment while symptomatic, and less or no impairment if adequate exposure controls are installed and are effective in reducing or eliminating symptoms. If effective exposure controls are not implemented, the same individual might have to be considered permanently impaired to work in that job, with recommendation for medical removal. If necessary, repeated assessment for long-term impairment/disability may be carried out two years after the exposure is reduced or terminated, when improvement of OA would be expected to have plateaued. If the patient continues to work, medical monitoring should be ongoing and reassessment of impairment/disability should be repeated as needed.
Workers who become disabled by OA or WAA may qualify for financial compensation for medical expenses and/or lost wages. In addition to directly reducing the financial impact of the disability on individual workers and their families, compensation may be necessary to provide proper medical treatment, initiate preventive intervention and obtain vocational rehabilitation. The worker’s and physician’s understanding of specific medico-legal issues may be important to ensuring that the diagnostic evaluation meets local requirements and does not result in compromise of the rights of the affected worker.
Although discussions of cost savings frequently focus on the inadequacy of compensation systems, genuinely reducing the financial and public health burden placed on society by OA and WAA will depend not only on improvements in compensation systems but, more importantly, on effectiveness of the systems deployed to identify and rectify, or prevent entirely, workplace exposures that are causing onset of new cases of asthma.
Conclusions
OA has become the most prevalent occupational respiratory disease in many countries. It is more common than generally recognized, can be severe and disabling, and is generally preventable. Early recognition and effective preventive interventions can substantially reduce the risk of permanent disability and the high human and financial costs associated with chronic asthma. For many reasons, OA merits more widespread attention among clinicians, health and safety specialists, researchers, health policy makers, industrial hygienists, and others interested in prevention of work-related diseases.
Diseases Caused by Organic Dusts
Organic Dust and Disease
Dusts of vegetable, animal and microbial origin have always been part of the human environment. When the first aquatic organisms moved to land some 450 million years ago, they soon developed defence systems against the many noxious substances present in the terrestrial environment, most of them of plant origin. Exposures to this environment usually cause no specific problems, even though plants contain a number of extremely toxic substances, particularly those present in or produced by moulds.
During the development of civilization, climatic conditions in some parts of the world necessitated certain activities to be undertaken indoors. Threshing in the Scandinavian countries was performed indoors during the winter, a practice mentioned by chroniclers in antiquity. The enclosure of dusty processes led to disease among the exposed persons, and one of the first published accounts of this is by the Danish bishop Olaus Magnus (1555, as cited by Rask-Andersen 1988). He described a disease among threshers in Scandinavia as follows:
“In separating the grain from the chaff, care must be taken to choose a time when there is a suitable wind which will sweep away the grain dust, so that it will not damage the vital organs of the threshers. This dust is so fine that it will almost unnoticeably penetrate into the mouth and accumulate in the throat. If this is not quickly dealt with by drinking fresh ale, the thresher may never again or only for a short period eat what he has threshed.”
With the introduction of machine processing of organic materials, treatment of large quantities of materials indoors with poor ventilation led to high levels of airborne dust. The descriptions by bishop Olaus Magnus and later by Ramazzini (1713) were followed by several reports on disease and organic dusts in the nineteenth century, particularly among cotton mill workers (Leach 1863; Prausnitz 1936). Later, the specific pulmonary disease common among farmers handling mouldy materials was also described (Campbell 1932).
During recent decades, a large number of reports on disease among persons exposed to organic dusts have been published. Initially, most of these were based on persons seeking medical help. The names of the diseases, when published, were often related to the particular environment where the disease was first recognized, and a bewildering array of names resulted, such as farmer’s lung, mushroom grower’s lung, brown lung and humidifier fever.
With the advent of modern epidemiology, more reliable figures were obtained for the incidence of occupational respiratory diseases related to organic dust (Rylander, Donham and Peterson 1986; Rylander and Peterson 1990). There was also advancement in the understanding of the pathological mechanisms underlying these diseases, particularly the inflammatory response (Henson and Murphy 1989). This paved the way for a more coherent picture of diseases caused by organic dusts (Rylander and Jacobs 1997).
The following will describe the different organic dust environments where disease has been reported, the disease entities themselves, the classical byssinosis disease and specific preventive measures.
Environments
Organic dusts are airborne particles of vegetable, animal or microbial origin. Table 1 lists examples of environments, work processes and agents involving the risk of exposure to organic dusts.
Table 1. Examples of sources of hazards of exposure to organic dust
Agriculture
Handling of grain, hay or other crops
Sugar-cane processing
Greenhouses
Silos
Animals
Swine/dairy confinement buildings
Poultry houses and processing plants
Laboratory animals, farm animals and pets
Waste-processing
Sewage water and silt
Household garbage
Composting
Industry
Vegetable fibre processing (cotton, flax, hemp, jute, sisal)
Fermentation
Timber and wood processing
Bakeries
Biotechnology processing
Buildings
Contaminated water in humidifiers
Microbial growth on structures or in ventilation ducts
Agents
It is now understood that the specific agents in the dusts are the major reason why disease develops. Organic dusts contain a multitude of agents with potential biological effects. Some of the major agents are found in table 2.
Table 2. Major agents in organic dusts with potential biological activity
Vegetable agents
Tannins
Histamine
Plicatic acid
Alkaloids (e.g., nicotine)
Cytochalasins
Animal agents
Proteins
Enzymes
Microbial agents
Endotoxins
(1→3)–β–D-glucans
Proteases
Mycotoxins
The relative role of each of these agents, alone or in combination with others, for the development of disease, is mostly unknown. Most of the information available relates to bacterial endotoxins which are present in all organic dusts.
Endotoxins are lipopolysaccharide compounds which are attached to the outer cell surface of Gram-negative bacteria. Endotoxin has a wide variety of biological properties. After inhalation it causes an acute inflammation (Snella and Rylander 1982; Brigham and Meyrick 1986). An influx of neutrophils (leukocytes) into the lung and the airways is the hallmark of this reaction. It is accompanied by activation of other cells and secretion of inflammatory mediators. After repeated exposures, the inflammation decreases (adaptation). The reaction is limited to the airway mucosa, and there is no extensive involvement of the lung parenchyma.
Another specific agent in organic dust is (1→3)-β-D-glucan. This is a polyglucose compound present in the cell wall structure of moulds and some bacteria. It enhances the inflammatory response caused by endotoxin and alters the function of inflammatory cells, particularly macrophages and T-cells (Di Luzio 1985; Fogelmark et al. 1992).
Other specific agents present in organic dusts are proteins, tannins, proteases and other enzymes, and toxins from moulds. Very little data are available on the concentrations of these agents in organic dusts. Several of the specific agents in organic dusts, such as proteins and enzymes, are allergens.
Diseases
The diseases caused by organic dusts are shown in table 3 with the corresponding International Classification of Disease (ICD) numbers (Rylander and Jacobs 1994).
Table 3. Diseases induced by organic dusts and their ICD codes
Bronchitis and pneumonitis (ICD J40)
Toxic pneumonitis (inhalation fever, organic dust toxic syndrome)
Airways inflammation (mucous membrane inflammation)
Chronic bronchitis (ICD J42)
Hypersensitivity pneumonitis (allergic alveolitis) (ICD J67)
Asthma (ICD J45)
Rhinitis, conjunctivitis
The primary route of exposure for organic dusts is by inhalation, and consequently the effects on the lung have received the major share of attention in research as well as in clinical work. There is, however, a growing body of evidence from published epidemiological studies and case reports as well as anecdotal reports, that systemic effects also occur. The mechanism involved seems to be a local inflammation at the target site, the lung, and a subsequent release of cytokines either with systemic effects (Dunn 1992; Michel et al. 1991) or an effect on the epithelium in the gut (Axmacher et al. 1991). Non-respiratory clinical effects are fever, joint pains, neurosensory effects, skin problems, intestinal disease, fatigue and headache.
The different disease entities as described in table 3 are easy to diagnose in typical cases, and the underlying pathology is distinctly different. In real life, however, a worker who has a disease due to organic dust exposure, often presents a mixture of the different disease entities. One person may have airways inflammation for a number of years, suddenly develop asthma and in addition have symptoms of toxic pneumonitis during a particularly heavy exposure. Another person may have subclinical hypersensitivity pneumonitis with lymphocytosis in the airways and develop toxic pneumonitis during a particularly heavy exposure.
A good example of the mixture of disease entities that may appear is byssinosis. This disease was first described in the cotton mills, but the individual disease entities are also found in other organic dust environments. An overview of the disease follows.
Byssinosis
The disease
Byssinosis was first described in the 1800s, and a classic report involving clinical as well as experimental work was given by Prausnitz (1936). He described the symptoms among cotton mill workers as follows:
“After working for years without any appreciable trouble except a little cough, cotton mill workers notice either a sudden aggravation of their cough, which becomes dry and exceedingly irritating¼ These attacks usually occur on Mondays ¼ but gradually the symptoms begin to spread over the ensuing days of the week; in time the difference disappears and they suffer continuously.”
The first epidemiological investigations were performed in England in the 1950s (Schilling et al. 1955; Schilling 1956). The initial diagnosis was based on the appearance of a typical Monday morning chest tightness, diagnosed using a questionnaire (Roach and Schilling 1960). A scheme for grading the severity of byssinosis based on the type and periodicity of symptoms was developed (Mekky, Roach and Schilling 1967; Schilling et al. 1955). Duration of exposure was used as a measure of dose and this was related to the severity of the response. Based on clinical interviews of large numbers of workers, this grading scheme was later modified to more accurately reflect the time intervals for the decrease in FEV1 (Berry et al. 1973).
In one study, a difference in the prevalence of byssinosis in mills processing different types of cotton was found (Jones et al. 1979). Mills using high-quality cotton to produce finer yarns had a lower prevalence of byssinosis than mills producing coarse yarns and using a lower quality of cotton. Thus in addition to exposure intensity and duration, both dose-related variables, the type of dust became an important variable for assessing exposure. Later it was demonstrated that the differences in the response of workers exposed to coarse and medium cottons was dependent not only on the type of cotton but on other variables that affect exposure, including: processing variables such as carding speed, environmental variables such as humidification and ventilation, and manufacturing variables such as different yarn treatments (Berry et al. 1973).
The next refinement of the relationship between exposure to cotton dust and a response (either symptoms or objective measures of pulmonary function), was the studies from the United States, comparing those who worked in 100% cotton to workers using the same cotton but in a 50:50 blend with synthetics and workers without exposure to cotton (Merchant et al. 1973). Workers exposed to 100% cotton had the highest prevalence of byssinosis independent of cigarette smoking, one of the confounders of exposure to cotton dust. This semiquantitative relationship between dose and response to cotton dust was further refined in a group of textile workers stratified by sex, smoking, work area and mill type. A relationship was observed in each of these categories between dust concentration in the lower dust ranges and byssinosis prevalence and/or change in forced expiratory volume in one second (FEV1).
In later investigations, the FEV1 decrease over the work shift has been used to assess the effects of exposure, and it is also a part of the US Cotton Dust Standard.
Byssinosis was long regarded as a peculiar disease with a mixture of different symptoms and no knowledge of the specific pathology. Some authors suggested that it was an occupational asthma (Bouhuys 1976). A workgroup meeting in 1987 analysed the symptomatology and pathology of the disease (Rylander et al. 1987). It was agreed that the disease comprised several clinical entities, generally related to organic dust exposure.
Toxic pneumonitis may appear the first time an employee works in the mill, particularly when working in the opening, blowing and carding sections (Trice 1940). Although habituation develops, the symptoms may reappear after an unusually heavy exposure later on.
Airways inflammation is the most widespread disease, and it appears at different degrees of severity from light irritation in the nose and airways to severe dry cough and breathing difficulties. The inflammation causes constriction of airways and a reduced FEV1. Airway responsiveness is increased as measured with a methacholine or histamine challenge test. It has been discussed whether airways inflammation should be accepted as a disease entity by itself or whether it merely represents a symptom. As the clinical findings in terms of severe cough with airways narrowing can lead to a decrease in work ability, it is justified to regard it as an occupational disease.
Continued airways inflammation over several years may develop into chronic bronchitis, particularly among heavily exposed workers in the blowing and carding areas. The clinical picture would be one of chronic obstructive pulmonary disease (COPD).
Occupational asthma develops in a small percentage of the workforce, but is usually not diagnosed in cross-sectional studies as the workers are forced to leave work because of the disease. Hypersensitivity pneumonitis has not been detected in any of the epidemiological studies undertaken, nor have there been case reports relating to cotton dust exposure. The absence of hypersensitivity pneumonitis may be due to the relatively low amount of moulds in cotton, as mouldy cotton is not acceptable for processing.
A subjective feeling of chest tightness, most common on Mondays, is the classical symptom of cotton dust exposure (Schilling et al. 1955). It is not, however, a feature unique to cotton dust exposure as it appears also among persons working with other kinds of organic dusts (Donham et al. 1989). Chest tightness develops slowly over a number of years but it can also be induced in previously unexposed persons, provided that the dose level is high (Haglind and Rylander 1984). The presence of chest tightness is not directly related to a decrease in FEV1.
The pathology behind chest tightness has not been explained. It has been suggested that the symptoms are due to an increased adhesiveness of platelets which accumulate in the lung capillaries and increase the pulmonary artery pressure. It is likely that chest tightness involves some kind of cell sensitization, as it takes repeated exposures for the symptom to develop. This hypothesis is supported by results from studies on blood monocytes from cotton workers (Beijer et al. 1990). A higher ability to produce procoagulant factor, indicative of cell sensitization, was found among cotton workers as compared to controls.
The environment
The disease was originally described among workers in cotton, flax and soft hemp mills. In the first phase of cotton treatment within the mills—bale opening, blowing and carding—more than half of the workers may have symptoms of chest tightness and airways inflammation. The incidence decreases as the cotton is processed, reflecting the successive cleaning of the causative agent from the fibre. Byssinosis has been described in all countries where investigations in cotton mills have been performed. Some countries like Australia have, however, unusually low incidence figures (Gun et al. 1983).
There is now uniform evidence that bacterial endotoxins are the causative agent for toxic pneumonitis and airways inflammation (Castellan et al. 1987; Pernis et al. 1961; Rylander, Haglind and Lundholm 1985; Rylander and Haglind 1986; Herbert et al. 1992; Sigsgaard et al. 1992). Dose-response relationships have been described and the typical symptoms have been induced by inhalation of purified endotoxin (Rylander et al. 1989; Michel et al. 1995). Although this does not exclude the possibility that other agents could contribute to the pathogenesis, endotoxins can serve as markers for disease risk. It is unlikely that endotoxins are related to the development of occupational asthma, but they could act as an adjuvant for potential allergens in cotton dust.
The case
The diagnosis of byssinosis is classically made using questionnaires with the specific question “Does your chest feel tight, and if so, on which day of the week?”. Persons with Monday morning chest tightness are classified as byssinotics according to a scheme suggested by Schilling (1956). Spirometry can be performed, and, according to the different combinations of chest tightness and decrease in FEV1, the diagnostic scheme illustrated in table 4 has evolved.
Table 4. Diagnostic criteria for byssinosis
Grade ½. Chest tightness on the first day of some working weeks
Grade 1. Chest tightness on the first day of every working week
Grade 2. Chest tightness on the first and other days of the working week
Grade 3. Grade 2 symptoms accompanied by evidence of permanent incapacity in the form of diminished effort intolerance and/or reduced ventilatory capacity
Treatment
Treatment in the light stages of byssinosis is symptomatic, and most of the workers learn to live with the slight chest tightness and bronchoconstriction that they experience on Mondays or when cleaning machinery or carrying out similar tasks with a higher than normal exposure. More advanced stages of airways inflammation or regular chest tightness several days of the week require transfer to less dusty operations. The presence of occupational asthma mostly requires work change.
Prevention
Prevention in general is dealt with in detail elsewhere in the Encyclopaedia. The basic principles for prevention in terms of product substitute, exposure limitation, worker protection and screening for disease apply also for cotton dust exposure.
Regarding product substitutes, it has been suggested that cotton with a low level of bacterial contamination be used. An inverse proof of this concept is found in reports from 1863 where the change to dirty cotton provoked an increase in the prevalence of symptoms among the exposed workers (Leach 1863). There is also the possibility of changing to other fibres, particularly synthetic fibres, although this is not always feasible from a product point of view. There is at present no production-applied technique to decrease the endotoxin content of cotton fibres.
Regarding dust reduction, successful programmes have been implemented in the United States and elsewhere (Jacobs 1987). Such programmes are expensive, and the costs for highly efficient dust removal may be prohibitive for developing countries (Corn 1987).
Regarding exposure control, the level of dust is not a sufficiently precise measure of exposure risk. Depending on the degree of contamination with Gram-negative bacteria and thus endotoxin, a given dust level may or may not be associated with a risk. For endotoxins, no official guidelines have been established. It has been suggested that a level of 200 ng/m3 is the threshold for toxic pneumonitis, 100 to 200 ng/m3 for acute airways constriction over the workshift and 10 ng/m3 for airways inflammation (Rylander and Jacobs 1997).
Knowledge about the risk factors and the consequences of exposure are important for prevention. The information basis has expanded rapidly during recent years, but much of it is not yet present in textbooks or other easily available sources. A further problem is that symptoms and findings in respiratory diseases induced by organic dust are non-specific and occur normally in the population. They may thus not be correctly diagnosed in the early stages.
Proper dissemination of knowledge concerning the effects of cotton and other organic dusts requires the establishment of appropriate training programmes. These should be directed not only towards workers with potential exposure but also towards employers and health personnel, particularly occupational health inspectors and engineers. Information must include source identification, symptoms and disease description, and methods of protection. An informed worker can more readily recognize work-related symptoms and communicate more effectively to a health care provider. Regarding health surveillance and screening, questionnaires are a major instrument to be used. Several versions of questionnaires specifically designed for diagnosing diseases induced by organic dust have been reported in the literature (Rylander, Peterson and Donham 1990; Schwartz et al. 1995). Lung function testing is also a useful tool for surveillance and diagnosis. Measurements of airway responsiveness have been found to be useful (Rylander and Bergström 1993; Carvalheiro et al. 1995). Other diagnostic tools such as measurements of inflammatory mediators or cell activity are still in the research phase.
Beryllium Disease
Beryllium disease is a systemic disorder involving multiple organs, with pulmonary manifestations being most prominent and common. It occurs on exposure to beryllium in its alloy form or in one of its various chemical compounds. Route of exposure is by inhalation and the disease can be either acute or chronic. Acute disease is extremely rare currently, and none has been reported since the first widespread industrial use of beryllium in the 1940s after industrial hygiene measures had been implemented to limit high-dose exposures. Chronic beryllium disease continues to be reported.
Beryllium, Alloys and Compounds
Beryllium, an industrial substance suspected of having carcinogenic potential, is notable for its lightness in weight, high tensile strength and corrosion resistance. Table 1 outlines the properties of beryllium and its compounds.
Table 1. Properties of beryllium and its compounds
|
Formula |
Specific |
Melting/boiling point (ºC) |
Solubility |
Description |
|
|
Beryllium (Be) |
9.01 (a.w.) |
1.85 |
1,298±5/2,970 |
— |
Grey to silver metal |
|
Beryllium oxide (BeO) |
25 |
3.02 |
2,530±30/— |
Soluble in acids and alkalis; insoluble in water |
White amorphous powder |
|
Beryllium fluoride1 (BeF2 ) |
47.02 |
1.99 |
Sublimes 800 °C |
Readily soluble in water; sparingly soluble in ethyl alcohol |
Hygroscopic solid |
|
Beryllium chloride2 (BeCl2 ) |
79.9 |
1.90 |
405/520 |
Very soluble in water; soluble in ethyl alcohol, benzene, ethyl ether and carbon disulphide |
White or slightly yellow deliquescent crystals |
|
Beryllium nitrate3 (Be(NO3 )2 ·3H2 O) |
187.08 |
1.56 |
60/142 |
Soluble in water and ethyl alcohol |
White to faintly yellow deliquescent crystals |
|
Beryllium nitride4 (Be3 N2 ) |
55.06 |
— |
2,200±100/— |
— |
Hard, refractory white crystals |
|
Beryllium sulphate |
177.2 |
1.71 |
100/— |
Soluble in water; insoluble in ethyl alcohol |
Colourless crystals |
1 Beryllium fluoride is made by the decompensation at 900–950 ºC of ammonium beryllium fluoride. Its main use is in the production of beryllium metal by reduction with magnesium.
2 Beryllium chloride is manufactured by passing chlorine over a mixture of beryllium oxide and carbon.
3 Beryllium nitrate is produced by the action of nitric acid on beryllium oxide. It is used as a chemical reagent and as a gas mantle hardener.
4 Beryllium nitride is prepared by heating beryllium metal powder in an oxygen-free, nitrogen atmosphere at 700–1,400 ºC. It is used in atomic energy reactions, including the production of the radioactive carbon isotope carbon-14.
5 Beryllium sulphate hydrate is produced by treating the fritted ore with concentrated suphuric acid.It is used in the production of metallic beryllium by the sulphate process.
Sources
Beryl (3BeO·Al2O3·6SiO2) is the chief commercial source of beryllium, the most abundant of the minerals containing high concentrations of beryllium oxide (10 to 13%). Major sources of beryl are to be found in Argentina, Brazil, India, Zimbabwe and the Republic of South Africa. In the United States, beryl is found in Colorado, South Dakota, New Mexico and Utah. Bertrandite, a low-grade ore (0.1 to 3%) with an acid-soluble beryllium content, is now being mined and processed in Utah.
Production
The two most important methods of extracting beryllium from the ore are the sulphate process and the fluoride process.
In the sulphate process, crushed beryl is melted in an arc furnace at 1,65°C and poured through a high-velocity water stream to form a frit. After heat treatment, the frit is ground in a ball mill and mixed with concentrated sulphuric acid to form a slurry, which is sprayed in the form of a jet into a directly heated, rotating sulphating mill. The beryllium, now in a water-soluble form, is leached from the sludge, and ammonium hydroxide is added to the leach liquor, which is then fed to a crystallizer where ammonium alum is crystallized out. Chelating agents are added to the liquor to hold iron and nickel in solution, sodium hydroxide is then added, and the sodium beryllate thus formed is hydrolyzed to precipitate beryllium hydroxide. The latter product may be converted to beryllium fluoride for reduction by magnesium to metallic beryllium, or to beryllium chloride for electrolytic reduction.
In the fluoride process (figure 1) a briquetted mixture of ground ore, sodium silicofluoride and soda ash is sintered in a rotating hearth furnace. The sintered material is crushed, milled and leached. Sodium hydroxide is added to the solution of beryllium fluoride thus obtained and the precipitate of beryllium hydroxide is filtered in a rotary filter. Metallic beryllium is obtained as in the previous process by the magnesium reduction of beryllium fluoride or by electrolysis of beryllium chloride.
Figure 1. Production of beryllium oxide by the fluoride process
Uses
Beryllium is used in alloys with a number of metals including steel, nickel, magnesium, zinc and aluminium, the most widely used alloy being beryllium-copper—properly called “a bronze”—which has a high tensile strength and a capacity for being hardened by heat treatment. Beryllium bronzes are used in non-spark tools, electrical switch parts, watch springs, diaphragms, shims, cams and bushings.
One of the largest uses of the metal is as a moderator of thermal neutrons in nuclear reactors and as a reflector to reduce the leakage of neutrons from the reactor core. A mixed uranium-beryllium source is often used as a neutron source. As a foil, beryllium is used as window material in x-ray tubes. Its lightness, high elastic modulus and heat stability make it an attractive material for the aircraft and aerospace industry.
Beryllium oxide is made by heating beryllium nitrate or hydroxide.
It is used in the manufacture of ceramics, refractory materials and other beryllium compounds. It was used for the manufacture of phosphors for fluorescent lamps until the incidence of beryllium disease in the industry caused its use for this purpose to be abandoned (in 1949 in the United States).
Hazards
Fire and health hazards are associated with processes involving beryllium. Finely divided beryllium powder will burn, the degree of combustibility being a function of particle size. Fires have occurred in dust filtration units and during the welding of ventilation ducting in which finely divided beryllium was present.
Beryllium and its compounds are highly toxic substances. Beryllium can affect all organ systems, although the primary organ involved is the lung. Beryllium causes systemic disease by inhalation and can distribute itself widely throughout the body after absorption from the lungs. Little beryllium is absorbed from the gastro-intestinal tract. Beryllium can cause skin irritation and its traumatic introduction into subcutaneous tissue can cause local irritation and granuloma formation.
Pathogenesis
Beryllium in all its forms, except for beryl ore, has been associated with disease. The route of entry is by inhalation and in the acute disease there is a direct toxic effect on both the nasopharyngeal mucosa and that of the entire tracheobronchial tree as well, causing oedema and inflammation. In the lung it causes an acute chemical pneumonitis. The major form of beryllium toxicity at this point in time is chronic beryllium disease. A beryllium-specific delayed type of hypersensitivity is the major pathway of chronic disease. The entry of beryllium into the system through the lungs leads to proliferation of specific CD+ lymphocytes, with beryllium acting as a specific antigen, either alone or as a hapten through an interleukin-2 (IL2) receptor pathway. Individual susceptibility to beryllium thus can be explained on the basis of the individual CD+ response. Release of lymphokines from the activated lymphocytes then can lead to granuloma formation and macrophage recruitment. Beryllium can be transported to sites outside the lung where it can cause granuloma formation. Beryllium is released slowly from different sites and it is excreted by the kidneys. This slow release can occur over a span of 20 to 30 years. The chronicity and latency of disease can probably be explained on the basis of the slow metabolism and release phenomenon. The immune mechanisms involved in the pathogenesis of beryllium disease also allow for specific approaches to diagnosis, which will be discussed below.
Histopathology
The primary pathological finding in beryllium disease is the formation of non-caseating granulomas in the lungs, lymph nodes and at other sites. Histopathological studies of lungs in patients with acute beryllium disease have shown a non-specific pattern of acute and subacute bronchitis and pneumonitis. In chronic beryllium disease, there are varying degrees of lymphocytic infiltration of the lung interstitium and non-caseating granuloma formation (figure 2).
Figure 2. Lung tissue in a patient with chronic beryllium disease
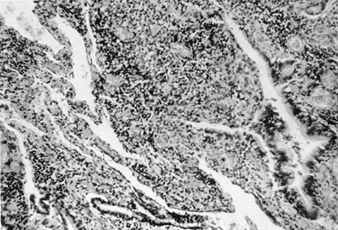
Both granulomas and round cell infiltration are visible
Many of the granulomas are located in the peribronchiolar areas. In addition, there can be histiocytes, plasma cells and giant cells with calcific inclusion bodies. If it is a case solely of granuloma formation, the long-term prognosis is better. The histology of the lung in chronic beryllium disease is indistinguishable from that of sarcoidosis. Non-caseating granulomas are also found in lymph nodes, liver, spleen, muscle and skin.
Clinical Manifestations
Skin injuries
Acid salts of beryllium cause allergic contact dermatitis. Such lesions may be erythematous, papular or papulovesicular, are commonly pruritic, and are found on exposed parts of the body. There is usually a delay of 2 weeks from first exposure to occurrence of the dermatitis, except in the case of heavy exposures, when an irritant reaction may be immediate. This delay is regarded as the time required to develop the hypersensitive state.
Accidental implantation of beryllium metal or crystals of a soluble beryllium compound in an abrasion, a crack in the skin or under the nail may cause an indurated area with central suppuration. Granulomas can also form at such sites.
Conjunctivitis and dermatitis may occur alone or together. In cases of conjunctivitis, periorbital oedema may be severe.
Acute disease
Beryllium nasopharyngitis is characterized by swollen and hyperaemic mucous membranes, bleeding points, fissures and ulceration. Perforation of the nasal septum has been described. Removal from exposure results in reversal of this inflammatory process within 3 to 6 weeks.
Involvement of the trachea and bronchial tree following exposure to higher levels of beryllium causes non-productive cough, substernal pain and moderate shortness of breath. Rhonchi and/or rales may be audible, and the x ray of the chest may show increased bronchovascular markings. The character and speed of onset and the severity of these signs and symptoms depend on the quality and quantity of exposure. Recovery is to be expected within 1 to 4 weeks if the worker is removed from further exposure.
The use of steroids is quite useful in countering the acute disease. No new cases of acute disease have been reported to the US Beryllium Case Registry in over 30 years. The Registry, which was started by Harriet Hardy in 1952, has almost 1,000 case records, among which are listed 212 acute cases. Almost all of these occurred in the fluorescent lamp manufacturing industry. Forty-four subjects with the acute disease subsequently developed chronic disease.
Chronic beryllium disease
Chronic beryllium disease is a pulmonary and systemic granulomatous disease caused by inhalation of beryllium. The latency of the disease can be from 1 to 30 years, most commonly occurring 10 to 15 years after first exposure. Chronic beryllium disease has a variable course with exacerbations and remissions in its clinical manifestations. However, the disease is usually progressive. There have been a few cases with chest x-ray abnormalities with a stable clinical course and without significant symptoms.
Exertional dyspnoea is the most common symptom of chronic beryllium disease. Other symptoms are cough, fatigue, weight loss, chest pain and arthralgias. Physical findings may be entirely normal or may include bibasilar crackles, lymphadenopathy, skin lesions, hepatosplenomegaly and clubbing. Signs of pulmonary hypertension may be present in severe, long-standing disease.
Renal stones and hyperuricaemia can occur in some patients and there have been rare reports of parotid gland enlargement and central nervous system involvement. The clinical manifestations of chronic beryllium disease are very similar to those of sarcoidosis.
Roentgenologic features
The x-ray pattern in chronic beryllium disease is non-specific and is similar to that which may be observed in sarcoidosis, idiopathic pulmonary fibrosis, tuberculosis, mycoses and dust disease (figure 3). Early in the course of the disease films may show granular, nodular or linear densities. These abnormalities may increase, decrease or remain unchanged, with or without fibrosis. Upper-lobe involvement is common. Hilar adenopathy, seen in approximately one-third of patients, is usually bilateral and accompanied by mottling of the lung fields. The absence of lung changes in the presence of adenopathy is a relative but not an absolute differential consideration in favour of sarcoidosis as opposed to chronic beryllium disease. Unilateral hilar adenopathy has been reported, but is quite rare.
Figure 3. Chest roentgenograph of a patient with chronic beryllium disease, showing diffuse fibronodular infiltrates and prominent hila
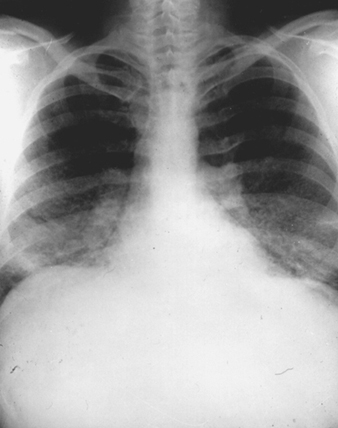
The x-ray picture does not correlate well with clinical status and does not reflect particular qualitative or quantitative aspects of the causal exposure.
Pulmonary function tests
Data from the Beryllium Case Registry show that 3 patterns of impairment may be found in chronic beryllium disease. Of 41 patients studied over a period of an average of 23 years after initial beryllium exposure, 20% had a restrictive defect, 36% had an interstitial defect (normal lung volumes and air flow rates but reduced diffusing capacity for carbon monoxide), 39% had an obstructive defect and 5% were normal. The obstructive pattern, which occurred in both smokers and non-smokers, was associated with granulomas in the peribronchial region. This study indicated that the pattern of impairment affects prognosis. Patients with interstitial defect fared best, with the least deterioration over a five-year interval. Patients with obstructive and restrictive defects experienced worsening of their impairment in spite of corticosteroid therapy.
Studies of lung function in beryllium extraction workers who were asymptomatic showed the presence of mild arterial hypoxaemia. This occurred usually within the first 10 years of exposure. In workers exposed to beryllium for 20 years or more there was a reduction in the forced vital capacity (FVC) and the forced expiratory volume in one second (FEV1). These findings suggest that the initial mild hypoxaemia could be due to the early alveolitis and that with further exposure and elapse of time the reduction in FEV1 and FVC could represent fibrosis and granuloma formation.
Other laboratory tests
Non-specific abnormal laboratory tests have been reported in chronic beryllium disease and include elevated sedimentation rate, erythrocytosis, increased gammaglobulin levels, hyperuricaemia and hypercalcaemia.
The Kveim skin test is negative in beryllium disease, whereas it may be positive in sarcoidosis. The angiotensin converting enzyme (ACE) level is usually normal in beryllium disease, but can be increased in 60% or more of patients with active sarcoidosis.
Diagnosis
Diagnosis of chronic beryllium disease for many years was based on the criteria developed through the Beryllium Case Registry, which included:
- a history of significant beryllium exposure
- evidence of lower respiratory tract disease
- abnormal chest x ray with interstitial fibronodular disease
- abnormal lung function tests with decreased carbon monoxide diffusing capacity (DLCO)
- pathological changes consistent with beryllium exposure in lung or thoracic lymph nodes
- the presence of beryllium in tissue.
Four of the six criteria had to be met and should have included either (1) or (6). Since the 1980s, advances in immunology have made it possible to make the diagnosis of beryllium disease without requiring tissue specimens for histological examination or beryllium analysis. The transformation of lymphocytes in blood in response to beryllium exposure (as in the lymphocyte transformation test, LTT) or lymphocytes from bronchoalveolar lavage (BAL) have been proposed by Newman et al. (1989) as useful diagnostic tools in making the diagnosis of beryllium disease in exposed subjects. Their data suggest that a positive blood LTT is indicative of sensitization. However, recent data show that the blood LTT does not correlate well with pulmonary disease. The BAL lymphocyte transformation correlates much better with abnormal pulmonary function and does not correlate well with concurrent abnormalities in the blood LTT. Thus, to make a diagnosis of beryllium disease, one needs a combination of clinical, radiological and lung function abnormalities and a positive LTT in the BAL. A positive blood LTT by itself is not diagnostic. Microprobe analysis of small tissue samples for beryllium is another recent innovation which could help in diagnosis of disease in small lung tissue samples obtained by transbronchial lung biopsy.
Sarcoidosis is the disorder most closely resembling chronic beryllium disease, and the differentiation may be difficult. Thus far, no cystic bone disease or involvement of the eye or tonsil has appeared in chronic beryllium disease. Similarly, the Kveim test is negative in beryllium disease. Skin testing to demonstrate beryllium sensitization is not recommended, in that the test itself is sensitizing, may possibly trigger systemic reactions in sensitized people and does not of itself establish that the presenting disease is necessarily beryllium related.
More sophisticated immunological approaches in differential diagnosis should allow for better differentiation from sarcoidosis in the future.
Prognosis
The prognosis of chronic beryllium disease has altered favourably during the years; it has been suggested that the longer delays in onset observed among beryllium workers may reflect lower exposure or lower beryllium body burden, resulting in a milder clinical course. Clinical evidence is that steroid therapy, if used when measurable disability first appears, in adequate doses for long enough periods, has improved the clinical status of many patients, allowing some of them to return to useful jobs. There is no clear evidence that steroids have cured chronic beryllium poisoning.
Beryllium and cancer
In animals, experimentally administered beryllium is a carcinogen, causing osteogenic sarcoma after intravenous injection in rabbits and lung cancer after inhalation in rats and monkeys. Whether beryllium may be a human carcinogen is a controversial issue. Some epidemiological studies have suggested an association, particularly after acute beryllium disease. This finding has been disputed by others. One can conclude that beryllium is carcinogenic in animals and there may be a link between lung cancer and beryllium in humans, particularly in those with the acute disease.
Safety and Health Measures
Safety and health precautions must cover the fire hazard as well as the much more serious toxicity danger.
Fire prevention
Arrangements must be made to prevent possible sources of ignition, such as the sparking or arcing of electrical apparatus, friction, and so forth, in the vicinity of finely divided beryllium powder. Equipment in which this powder has been present should be emptied and cleaned before acetylene or electrical welding apparatus is used on it. Oxide-free, ultrafine beryllium powder that has been prepared in inert gas is liable to ignite spontaneously on exposure to air.
Suitable dry powder—not water—should be used to extinguish a beryllium fire. Full personal protective equipment, including respiratory protective equipment, should be worn and firefighters should bathe afterwards and arrange for their clothing to be laundered separately.
Health protection
Beryllium processes must be conducted in a carefully controlled manner to protect both the worker and the general population. The main risk takes the form of airborne contamination and the process and plant should be designed to give rise to as little dust or fume as possible. Wet processes should be used instead of dry processes, and the ingredients of beryllium-containing preparations should be unified as aqueous suspensions instead of as dry powders; whenever possible the plant should be designed as groups of separate enclosed units. The permissible concentration of beryllium in the atmosphere is so low that enclosure must be applied even to wet processes, otherwise escaping splashes and spills can dry out and the dust can enter the atmosphere.
Operations from which dust may be evolved should be conducted in areas with maximum degree of enclosure consistent with the needs of manipulation. Some operations are performed in glove boxes, but many more are conducted in enclosures provided with exhaust ventilation similar to that installed in chemical fume cupboards. Machining operations may be ventilated by high-velocity, low-volume local exhaust systems or by hooded enclosures with exhaust ventilation.
To check the effectiveness of these precautionary measures atmosphere monitoring should be done in such a manner that the daily average exposure of workers to respirable beryllium can be calculated. The work area should be cleaned regularly by means of a proper vacuum cleaner or a wet mop. Beryllium processes should be segregated from the other operations in the factory.
Personal protective equipment should be provided for workers engaged in beryllium processes. Where they are fully employed in processes involving the manipulation of beryllium compounds or in processes associated with the extraction of the metal from the ore, provision should be made for a complete change of clothing so that the workers do not go home wearing clothing in which they have been working. Arrangements should be made for the safe laundering of such working clothes, and protective overalls should be provided even to laundry workers to ensure that they too are not exposed to risk. These arrangements should not be left to normal home laundering procedures. Cases of beryllium poisoning in the families of workers have been attributed to workers taking contaminated clothing home or wearing them in the home.
An occupational health standard of 2μg/m3, proposed in 1949 by a committee operating under the auspices of the US Atomic Energy Commission, continues to be widely observed. Existing interpretations generally permit fluctuations to a “ceiling” of 5μg/m3 as long as the time-weighted average is not exceeded. Additionally, an “acceptable maximum peak above the ceiling concentration for an eight-hour shift” of 25μg/m3 for up to 30 min is also permissible. These operational levels are achievable in current industrial practice, and there is no evidence of adverse health experience among persons working in an environment thus controlled. Because of a possible link between beryllium and lung cancer it has been suggested that the allowable limit be reduced to 1μg/m3, but no official action has been taken on this suggestion in the United States.
The population at risk for developing beryllium disease is that which in some manner deals with beryllium in its extraction or subsequent use. However, a few “neighbourhood” cases have been reported from a distance 1 to 2 km from beryllium extraction plants.
Pre-employment and periodical medical examinations of workers exposed to beryllium and its compounds are compulsory in a number of countries. Recommended evaluation includes an annual respiratory questionnaire, a chest x ray and lung function tests. With advances in immunology, the LTT may also become a routine evaluation, although at this time not enough data are available to recommend its use routinely. With evidence of beryllium disease, it is unwise to allow a worker to be exposed to beryllium further, even though the workplace meets the threshold criteria for beryllium concentration in the air.
Treatment
The major step in therapy is avoidance of further exposure to beryllium. Corticosteroids are the primary mode of therapy in chronic beryllium disease. Corticosteroids appear to alter the course of disease favourably but do not “cure” it.
Corticosteroids should be started on a daily basis with a relatively high dose of Prednisone of 0.5 to 1 mg per kg or more, and continued until improvement occurs or no further deterioration in clinical or lung function tests occurs. Usually this takes 4 to 6 weeks. Slow reduction of steroids is recommended, and eventually alternate-day therapy may be possible. Steroid therapy ordinarily becomes a lifelong necessity.
Other supportive measures such as supplemental oxygen, diuretics, digitalis and antibiotics (when infection exists) are indicated as the clinical condition of the patient would dictate. Immunization against influenza and pneumococcus should also be considered, as with any patient with chronic respiratory disease.
Pneumoconioses: Definition
The expression pneumoconiosis, from the Greek pneuma (air, wind) and konis (dust) was coined in Germany by Zenker in 1867 to denote changes in the lungs caused by the retention of inhaled dust. Gradually, the need for distinction between the effects of various types of dust became evident. It was necessary to discriminate among mineral or vegetable dust and their microbiological component. Consequently, the Third International Conference of Experts on Pneumoconiosis, organized by the ILO in Sydney in 1950, adopted the following definition: “Pneumoconiosis is a diagnosable disease of the lungs produced by the inhalation of dust, the term ‘dust’ being understood to refer to particulate matter in the solid phase, but excluding living organisms.”
However, the word disease seems to imply some degree of health impairment which may not be the case with pneumoconioses not connected with the development of lung fibrosis/scarring. In general, the reaction of lung tissue to the presence of dust varies with different dusts. Non-fibrogenic dusts evoke a tissue reaction in lungs characterized by minimal fibrotic reaction and absence of lung function impairment. Such dusts, examples of which are finely divided dusts of kaolinite, titanium dioxide, stannous oxide, barium sulphate and ferric oxide, are frequently referred to as biologically inert.
Fibrogenic dust such as silica or asbestos causes a more pronounced fibrogenic reaction resulting in scars in the lung tissue and obvious disease. The division of dusts into fibrogenic and non-fibrogenic varieties is by no means sharp because there are many minerals, notably silicates, which are intermediate in their ability to produce fibrotic lesions in the lungs. Nevertheless, it proved useful for clinical purposes and is reflected in the classification of pneumoconioses.
A new definition of pneumoconioses was adopted at the Fourth International Conference on Pneumoconiosis, Bucharest, 1971: “Pneumoconiosis is the accumulation of dust in the lungs and the tissue reactions to its presence. For the purpose of this definition, ‘dust’ is meant to be an aerosol composed of solid inanimate particles.”
In order to avoid any misinterpretation, the expression non-neoplastic is sometimes added to the words “tissue reaction”.
The Working Group at the Conference made the following comprehensive statement:
The Definition of Pneumoconiosis
Earlier on, in 1950, a definition of pneumoconiosis was established at the 3rd International Conference of Experts on Pneumoconiosis and this has continued to be used until the present time. In the meantime, the development of new technologies has resulted in more occupational risks, particularly those related to the inhalation of airborne contaminants. Increased knowledge in the field of occupational medicine has enabled new pulmonary diseases of occupational origin to be recognized but has also demonstrated the necessity for a re-examination of the definition of pneumoconiosis established in 1950. The ILO therefore arranged for a Working Group to be convened within the framework of the IVth International Pneumoconiosis Conference in order to examine the question of the definition of pneumoconiosis. The Working Group held a general discussion on the matter and proceeded to examine a number of proposals submitted by its members. It finally adopted a new definition of pneumoconiosis which was prepared together with a commentary. This text is reproduced below.
In recent years a number of countries have included under pneumoconiosis, because of socio-economic reasons, conditions which are manifestly not pneumoconiosis, but are nevertheless occupational pulmonary diseases. Under the term “disease” are included for preventive reasons the earliest manifestations which are not necessarily disabling or life shortening. Therefore the Working Group has undertaken to redefine pneumoconiosis as the accumulation of dust in the lungs and the tissue reactions to its presence. For the purpose of this definition, “dust” is meant to be an aerosol composed of solid inanimate particles. From a pathological point of view pneumoconiosis may be divided for the sake of convenience into collagenous or non-collagenous forms. A non-collagenous pneumoconiosis is caused by a non-fibrogenic dust and has the following characteristics:
- the alveolar architecture remains intact
- the stromal reaction is minimal and consists mainly of reticulin fibres
- the dust reaction is potentially reversible.
Examples of non-collagenous pneumoconiosis are those caused by pure dusts of tin oxide (stannosis) and barium sulphate (barytosis).
Collagenous pneumoconiosis is characterised by:
- permanent alteration or destruction of alveolar architecture
- collagenous stromal reaction of moderate to maximal degree, and
- permanent scarring of lung.
Such collagenous pneumoconiosis may be caused by fibrogenic dusts or by an altered tissue response to a non-fibrogenic dust.
Examples of collagenous pneumoconiosis caused by fibrogenic dusts are silicosis and asbestosis, whereas complicated coalworkers’ pneumoconiosis or progressive massive fibrosis (PMF) is an altered tissue response to a relatively non-fibrogenic dust. In practice, the distinction between collagenous and non-collagenous pneumoconiosis is difficult to establish. Continued exposure to the same dust, such as coal dust, may cause transition from a non-collagenous to a collagenous form. Furthermore, exposure to a single dust is now becoming less common and exposures to mixed dusts having different degrees of fibrogenic potential may result in pneumoconiosis which can range from the non-collagenous to the collagenous forms. There are in addition occupational chronic pulmonary diseases which, although they develop from the inhalation of dust are excluded from the pneumoconiosis because the particles are not known to accumulate in the lungs. The following are examples of potentially disabling occupational chronic pulmonary diseases: byssinosis, berylliosis, farmers’ lung, and related diseases. They have one common denominator, namely the aetiologic component of dust has sensitized the pulmonary or bronchial tissue so that if the lung tissue responds, the inflammation tends to be granulomatous and if the bronchial tissue responds, there is apt to be bronchial constriction. Exposures to noxious inhaled materials in certain industries are associated with an increased risk of mortality from carcinoma of the respiratory tract. Examples of such materials are radioactive ores, asbestos and chromates.
Adopted at the IVth ILO International Conference on Pneumoconiosis. Bucharest, 1971.
ILO International Classification of Radiographs of Pneumoconioses
Despite all the national and international energies devoted to their prevention, pneumoconioses are still very present both in industrialized and developing countries, and are responsible for the disability and impairment of many workers. This is why the International Labour Office (ILO), the World Health Organization (WHO) and many national institutes for occupational health and safety continue their fight against these diseases and to propose sustainable programmes for preventing them. For instance, the ILO, the WHO and the US National Institute for Occupational Safety and Health (NIOSH) have proposed in their programmes to work in cooperation on a global fight against silicosis. Part of this programme is based on medical surveillance which includes the reading of thoracic radiographs to help diagnose this pneumoconiosis. This is one example which explains why the ILO, in cooperation with many experts, has developed and updated on a continuous basis a classification of radiographs of pneumoconioses that provides a means for recording systematically the radiographic abnormalities in the chest provoked by the inhalation of dust. The scheme is designed for classifying the appearances of posterio-anterior chest radiographs.
The object of the classification is to codify the radiographic abnormalities of pneumoconioses in a simple, reproducible manner. The classification does not define pathological entities, nor take into account working capacity. The classification does not imply legal definitions of pneumoconioses for compensation purposes, nor imply a level at which compensation is payable. Nevertheless, the classification has been found to have wider uses than anticipated. It is now extensively used internationally for epidemiological research, for the surveillance of those industry occupations and for clinical purposes. Use of the scheme may lead to better international comparability of pneumoconioses statistics. It is also used for describing and recording, in a systematic way, part of the information needed for assessing compensation.
The most important condition for using this system of classification with full value from a scientific and ethical point of view is to read, at all times, films to be classified by systematically referring to the 22 standard films provided in the ILO International Classification set of standard films. If the reader attempts to classify a film without referring to any of the standard films, then no mention of reading according to the ILO International Classification of Radiographs should be made. The possibility of deviating from the classification by over or under reading is so risky that his or her reading should not be used at least for epidemiological research or international comparability of pneumoconioses statistics.
The first classification was proposed for silicosis at the First International Conference of Experts on Pneumoconioses, held in Johannesburg in 1930. It combined both radiographic appearances and impairment of lung functions. In 1958, a new classification based purely on radiographic changes was established (Geneva classification 1958). Since, it has been revised several times, the last time in 1980, always with the objective of providing improved versions to be extensively used for clinical and epidemiological purposes. Each new version of the classification promoted by the ILO has brought modifications and changes based on international experience gained in the use of earlier classifications.
In order to provide clear instructions for the use of the classification, the ILO issued in 1970 a publication entitled International Classification of Radiographs of Pneumoconioses/1968 in the Occupational Safety and Health Series (No. 22). This publication was revised in 1972 as ILO U/C International Classification of Radiographs of Pneumoconioses/1971 and again in 1980 as Guidelines for the use of ILO International Classification of Radiographs of Pneumoconioses, revised edition 1980. The description of standard radiographs is given in table 1.
Table 1. Description of standard radiographs
| 1980 Standard radiographs showing | Small opacities | Pleural thickening | ||||||||||
| Chest wall | ||||||||||||
| Technical quality | Profusion | Shape- size | Extent | Large opacities | Circum- scribed (plaques) | Diffuse | Diaphragm | Costo- phrenic angle obliteration | Pleural calcification | Symbols | Comments | |
| 0/0 (example 1) | 1 | 0/0 | – | – | No | No | No | No | No | No | None | Vascular pattern is well illustrated |
| 0/0 (example 2) | 1 | 0/0 | – | – | No | No | No | No | No | No | None | Also shows vascular pattern, but not as clearly as example 1 |
| 1/1; p/p | 1 | 1/1 | p/p | R L x x x x x x | A | No | No | No | No | No | rp. | Rheumatoid pneumoconiosis in left lower zone. Small opacities are present in all zones, but the profusion in the right-upper zone is typical of (some would say a little more profuse than) that classifiable as category 1/1 |
| 2/2; p/p | 2 | 2/2 | p/p | R L x x x x x x | No | No | No | No | No | No | pi; tb. | Quality defect: radiograph is too light |
| 3/3; p/p | 1 | 3/3 | p/p | R L x x x x x x | No | No | No | No | Yes R L x – | No | ax. | None |
| 1/1; q/q | 1 | 1/1 | q/q | R L x x x x – – | No | No | No | No | No | No | None | Illustrates profusion 1/1 better than shape or size |
| 2/2; q/q | 1 | 2/2 | q/q | R L x x x x x x | No | No | Yes R L x x width: a a extent: 1 1 | No | Yes R L x x | No | None | None |
| 3/3; q/q | 2 | 3/3 | q/q | R L x x x x x x | No | No | No | No | No | No | pi. | Quality defects: poor definition of pleura and cut basal angles |
| 1/1; r/r | 2 | 1/1 | r/r | R L x x x x – – | No | No | No | No | Yes R L – x | No | None | Quality defect: subject movement. Profusion of small opacities is more marked in right lung |
| 2/2; r/r | 2 | 2/2 | r/r | R L x x x x x x | No | No | No | No | No | No | None | Quality defects: radiograph too light and contrast too high. The heart shadow is slightly displaced to the left |
| 3/3; r/r | 1 | 3/3 | r/r | R L x x x x x x | No | No | No | No | No | No | ax; ih. | None |
| 1/1; s/t | 2 | 1/1 | s/t | R L x – x x x x | No | No | No | No | No | No | kl. | Quality defect: cut bases. Kerley lines in lower right zone |
| 2/2; s/s | 2 | 2/2 | s/s | R L – – x x x x | No | No | No | No | No | No | em. | Quality defect: distortion of bases due to shrinking. Emphysema in upper zones |
| 3/3; s/s | 2 | 3/3 | s/s | R L x x x x x x | No | No | Yes R L x x width: a a extent: 3 3 | No | No | No | ho; ih; pi. | Quality defect: radiograph is too light. Honeycomb lung appearance is not marked |
| 1/1; t/t Costophrenic angle obliteration | 1 | 1/1 | t/t | R L – – x x x x | No | No | Yes R L x x width: a a extent: 2 2 | No | Yes R L x – | Yes R L – x extent: 2 | None | This radiograph defines the lower limit for costophrenic angle obliteration. Note shrinkage in lower lung fields |
| 2/2; t/t | 1 | 2/2 | t/t | R L x x x x x x | No | No | Yes R L x x width: a a extent: 1 1 | No | No | No | ih. | Pleural thickening is present in the apices of the lung |
| 3/3; t/t | 1 | 3/3 | t/t | R L x x x x x x | No | No | No | No | No | No | hi; ho; id; ih; tb. | None |
| 1/1; u/u 2/2; u/u 3/3; u/u | – | – | – | – | – | – | – | – | – | – | – | This composite radiograph illustrates the mid-categories of profusion of small opacities classifiable for shape and size as u/u. |
| A | 2 | 2/2 | p/q | R L x x x x x x | A | No | No | No | No | No | No | Quality defects: radiograph is too light and pleural definition is poor |
| B | 1 | 1/2 | p/q | R L x x x x x x | B | No | No | No | No | No | ax; co. | Definition of pleura is slightly imperfect |
| C | 1 | 2/1 | q/t | R L x x x x x x | C | No | No | No | No | No | bu; di; em; es; hi; ih. | The small opacities are difficult to classify because of the presence of the large opacities. Note the left costophrenic angle obliteration. This is not classifiable because it does not reach the lower limit defined by the standard radiograph 1/1; t/t |
| Pleural thickening (circumscribed) | – | – | – | – | – | Yes | No | No | No | No | The pleural thickening present face on, is of indeterminate width, and extent 2 | |
| Pleural thickening (diffuse) | – | – | – | – | – | No | Yes | No | No | Yes | The pleural thickening present in profile, is of width a, and extent 2. Not associated small calcifications | |
| Pleural thickening (calcification) diaphragm | – | – | – | – | – | No | No | Yes | No | Yes | Circumscribed, calcified pleural thickening of extent 2 | |
| Pleural thickening (calcification) chest wall | – | – | – | – | – | Yes | No | No | No | Yes | Calcified and uncalcified pleural thickening present face on, is of indeterminate width, and extent 2 | |
ILO 1980 Classification
The 1980 revision was carried out by the ILO with the cooperation of the Commission of the European Communities, NIOSH and the American College of Radiology. The summary of the classification is given in table 2. It retained the principle of former classifications (1968 and 1971).
Table 2. ILO 1980 International Classification of Radiographs of Pneumoconioses: Summary of details of classification
| Features | Codes | Definitions | |
| Technical quality | |||
| 1 | Good. | ||
| 2 | Acceptable, with no technical defect likely to impair classification of the radiograph of pneumoconiosis. | ||
| 3 | Poor, with some technical defect but still acceptable for classification purposes. | ||
| 4 | Unacceptable. | ||
| Parenchymal abnormalities | |||
| Small opacities | Profusion | The category of profusion is based on assessment of the concentration of opacities by comparison with the standard radiographs. | |
| 0/- 0/0 0/1 1/0 1/1 1/2 2/1 2/2 2/3 3/2 3/3 3/+ | Category O—small opacities absent or less profuse than the lower limit of category 1. Categories 1, 2 and 3—increasing profusion of small opacities as defined by the corresponding standard radiographs. | ||
| Extent | RU RM RL LU LM LL | The zones in which the opacities are seen are recorded. The right (R) and left (L) thorax are both divided into three zones—upper (U), middle (M) and lower (L). The category of profusion is determined by considering the profusion as a whole over the affected zones of the lung and by comparing this with the standard radiographs. | |
| Shape and Size | |||
| Rounded | p/p q/q r/r | The letters p, q and r denote the presence of small, rounded opacities. Three sizes are defined by the appearances on standard radiographs: p = diameter up to about 1.5 mm q = diameter exceeding about 1.5 mm and up to about 3 mm r = diameter exceeding about 3 mm and up to about 10 mm | |
| Irregular | s/s t/t u/u | The letters s, t and u denote the presence of small, irregular opacities. Three sizes are defined by the appearances on standard radiographs: s = width up to about 1.5 mm t = width exceeding about 1.5 mm and up to about 3 mm u = width exceeding 3 mm and up to about 10 mm | |
| Mixed | p/s p/t p/u p/q p/r q/s q/t q/u q/p q/r r/s r/t r/u r/p r/q s/p s/q s/r s/t s/u t/p t/q t/r t/s t/u u/p u/q u/r u/s u/t | For mixed shapes (or sizes) of small opacities, the predominant shape and size is recorded first. The presence of a significant number of another shape and size is recorded after the oblique stroke. | |
| Large opacities | A B C | The categories are defined in terms of the dimensions of the opacities. Category A – an opacity having a greatest diameter exceeding about 10 mm and up to and including 50 mm, or several opacities each greater than about 10 mm, the sum of whose greatest diameters does not exceed about 50 mm. Category B – one or more opacities larger or more numerous than those in category A whose combined area does not exceed the equivalent of the right upper zone. Category C – one or more opacities whose combined area exceeds the equivalent of the right upper zone. | |
| Pleural abnormalities | |||
| Pleural thickening | |||
| Chest wall | Type | Two types of pleural thickening of the chest wall are recognized: circumscribed (plaques) and diffuse. Both types may occur together | |
| Site | R L | Pleural thickening of the chest wall is recorded separately for the right (R) and left (L) thorax. | |
| Width | a b c | For pleural thickening seen along the lateral chest wall the measurement of maximum width is made from the inner line of the chest wall to the inner margin of the shadow seen most sharply at the parenchymal-pleural boundary. The maximum width usually occurs at the inner margin of the rib shadow at its outermost point. a = maximum width up to abut 5 mm b = maximum width over about 5 mm and up to about 10 mm c = maximum width over about 10 mm | |
| Face on | Y N | The presence of pleural thickening seen face-on is recorded even if it can be seen also in profile. If pleural thickening is seen face-on only, width cannot usually be measured. | |
| Extent | 1 2 3 | Extent of pleural thickening is defined in terms of the maximum length of pleural involvement, or as the sum of maximum lengths, whether seen in profile or face-on. 1 = total length equivalent up to one quarter of the projection of the lateral chest wall 2 = total length exceeding one quarter but not one half of the projection of the lateral chest wall 3 = total length exceeding one half of the projection of the lateral chest wall | |
| Diaphragm | Presence | Y N | A plaque involving the diaphragmatic pleura is recorded as present (Y) or absent (N), separately for the right (R) and left (L) thorax. |
| Site | R L | ||
| Costrophrenic angle obliteration | Presence | Y N | The presence (Y) or absence (N) of costophrenic angle obliteration is recorded separately from thickening over other areas, for the right (R) and left (L) thorax. The lower limit for this obliteration is defined by a standard radiograph |
| Site | R L | If the thickening extends up the chest wall, then both costophrenic angle obliteration and pleural thickening should be recorded. | |
| Pleural calcification | Site | The site and extent of pleural calcification are recorded separately for the two lungs, and the extent defined in terms of dimensions. | |
| Chest wall | R L | ||
| Diaphragm | R L | ||
| Other | R L | “Other” includes calcification of the mediastinal and pericardial pleura. | |
| Extent | 1 2 3 | 1 = an area of calcified pleura with greatest diameter up to about 20 mm, or a number of such areas the sum of whose greatest diameters does not exceed about 20 mm. 2 = an area of calcified pleura with greatest diameter exceeding about 20 mm and up to about 100 mm, or a number of such areas the sum of whose greatest diameters exceeds about 20 mm but does not exceed about 100 mm. 3 = an area of calcified pleura with greatest diameter exceeding about 100 mm, or a number of such areas whose sum of greatest diameters exceeds about 100 mm. | |
| Symbols | |||
| It is to be taken that the definition of each of the symbols is preceded by an appropriate word or phrase such as “suspect”, “changes suggestive of”, or “opacities suggestive of”, etc. | |||
| ax | Coalescence of small pneumoconiotic opacities | ||
| bu | Bulla(e) | ||
| ca | Cancer of lung or pleura | ||
| cn | Calcification in small pneumoconiotic opacities | ||
| co | Abnormality of cardiac size or shape | ||
| cp | Cor pulmonale | ||
| cv | Cavity | ||
| di | Marked distortion of the intrathoracic organs | ||
| ef | Effusion | ||
| em | Definite emphysema | ||
| es | Eggshell calcification of hilar or mediastinal lymph nodes | ||
| fr | Fractured rib(s) | ||
| hi | Enlargement of hilar or mediastinal lymph nodes | ||
| ho | Honeycomb lung | ||
| id | Ill-defined diaphragm | ||
| ih | Ill-defined heart outline | ||
| kl | Septal (Kerley) lines | ||
| od | Other significant abnormality | ||
| pi | Pleural thickening in the interlobar fissure of mediastinum | ||
| px | Pneumothorax | ||
| rp | Rheumatoid pneumoconiosis | ||
| tb | Tuberculosis | ||
| Comments | |||
| Presence | Y N | Comments should be recorded pertaining to the classification of the radiograph, particularly if some other cause is thought to be responsible for a shadow which could be thought by others to have been due to pneumoconiosis; also to identify radiographs for which the technical quality may have affected the reading materially. | |
The Classification is based on a set of standard radiographs, a written text and a set of notes (OHS No. 22). There are no features to be seen in a chest radiograph which are pathognomonic of dust exposure. The essential principle is that all appearances which are consistent with those defined and represented in the standard radiographs and the guideline for the use of the ILO International Classification, are to be classified. If the reader believes that any appearance is probably or definitively not dust related, the radiograph should not be classified but an appropriate comment must be added. The 22 standard radiographs have been selected after international trials, in such a way as to illustrate the mid-categories standards of profusion of small opacities and to give examples of category A, B and C standards for large opacities. Pleural abnormalities (diffuse pleural thickening, plaques and obliteration of costophrenic angle) are also illustrated on different radiographs.
Discussion in particular at the Seventh International Pneumoconioses Conference, held in Pittsburgh in 1988, indicated the need for improvement of some parts of the classification, in particular those concerning pleural changes. A discussion group meeting on the revision of the ILO International Classification of Radiographs of Pneumoconioses was convened in Geneva by the ILO in November 1989. The experts made the suggestion that the short classification is of no advantage and can be deleted. As regards pleural abnormalities, the group agreed that this classification would now be divided into three parts: “Diffuse pleural thickening”; “Pleural plaques”; and “Costophrenic angle obliteration”. Diffuse pleural thickening might be divided into chest wall and diaphragm. They were identified according to the six zones—upper, middle and lower, of both right and left lungs. If a pleural thickening is circumscribed, it could be identified as a plaque. All plaques should be measured in centimetres. The obliteration of the costophrenic angle should be systematically noted (whether it exists or not). It is important to identify whether the costophrenic angle is visible or not. This is because of its special importance in relation to pleural diffuse thickening. Whether plaques are classified or not should be merely indicated by a symbol. The flattening of the diaphragm should be recorded by an additional symbol since it is a very important feature in asbestos exposure. The presence of plaques should be recorded in these boxes using the appropriate symbol “c” (calcified) or “h” (hyaline).
A full description of the classification, including its applications and limitation is found in the publication (ILO 1980). The revision of the classification of radiographs is a continuous ILO process, and a revised guideline should be published in the near future (1997-98) taking into account the recommendations of these experts.
Aetiopathogensis of pneumoconioses
Pneumoconioses have been recognized as occupational diseases for a long time. Substantial efforts have been directed to research, primary prevention and medical management. But physicians and hygienists report that the problem is still present in both industrialized and industrializing countries (Valiante, Richards and Kinsley 1992; Markowitz 1992). As there is strong evidence that the three main industrial minerals responsible for the pneumoconioses (asbestos, coal and silica) will continue to have some economical importance, thus further entailing possible exposure, it is expected that the problem will continue to be of some magnitude throughout the world, particularly among underserved populations in small industries and small mining operations. Practical difficulties in primary prevention, or insufficient understanding of the mechanisms responsible for the induction and the progression of the disease are all factors which could possibly explain the continuing presence of the problem.
The aetiopathogenesis of pneumoconioses can be defined as the appraisal and understanding of all the phenomena occurring in the lung following the inhalation of fibrogenic dust particles. The expression cascade of events is often found in the literature on the subject. The cascade is a series of events that first exposure and at its farthest extent progresses to the disease in its more severe forms. If we except the rare forms of accelerated silicosis, which can develop after only a few months of exposure, most of the pneumoconioses develop following exposure periods measured in decades rather than years. This is especially true nowadays in workplaces adopting modern standards of prevention. Aetiopathogenesis phenomena should thus be analysed in terms of its long-term dynamics.
In the last 20 years, a large amount of information has become available on the numerous and complex pulmonary reactions involved in interstitial lung fibrosis induced by several agents, including mineral dusts. These reactions were described at the biochemical and cellular level (Richards, Masek and Brown 1991). Contributions were made by not only physicists and experimental pathologists but also by clinicians who used bronchoalveolar lavage extensively as a new pulmonary technique of investigation. These studies pictured aetiopathogenesis as a very complex entity, which can nonetheless be broken down to reveal several facets: (1) the inhalation itself of dust particles and the consequent constitution and significance of the pulmonary burden (exposure-dose-response relationships), (2) the physicochemical characteristics of the fibrogenic particles, (3) biochemical and cellular reactions inducing the fundamental lesions of the pneumoconioses and (4) the determinants of progression and complication. The later facet must not be ignored, since the more severe forms of pneumoconioses are the ones which entail impairment and disability.
A detailed analysis of the aetiopathogenesis of the pneumoconioses is beyond the scope of this article. One would need to distinguish the several types of dust and to go deeply into numerous specialized areas, some of which are still the subject of active research. But interesting general notions emerge from the currently available amount of knowledge on the subject. They will be presented here through the four “facets” previously mentioned and the bibliography will refer the interested reader to more specialized texts. Examples will be essentially given for the three main and most documented pneumoconioses: asbestosis, coal workers’ pneumoconioses (CWP) and silicosis. Possible impacts on prevention will be discussed.
Exposure-Dose-Response Relationships
Pneumoconioses result from the inhalation of certain fibrogenic dust particles. In the physics of aerosols, the term dust has a very precise meaning (Hinds 1982). It refers to airborne particles obtained by mechanical comminution of a parent material in a solid state. Particles generated by other processes should not be called dust. Dust clouds in various industrial settings (e.g., mining, tunnelling, sand blasting and manufacturing) generally contain a mixture of several types of dust. The airborne dust particles do not have a uniform size. They exhibit a size distribution. Size and other physical parameters (density, shape and surface charge) determine the aerodynamic behaviour of the particles and the probability of their penetration and deposition in the several compartments of the respiratory system.
In the field of pneumoconioses, the site compartment of interest is the alveolar compartment. Airborne particles small enough to reach this compartments are referred to as respirable particles. All particles reaching the alveolar compartments are not systematically deposited, some being still present in the exhaled air. The physical mechanisms responsible for deposition are now well understood for isometric particles (Raabe 1984) as well as for fibrous particles (Sébastien 1991). The functions relating the probability of deposition to the physical parameters have been established. Respirable particles and particles deposited in the alveolar compartment have slightly different size characteristics. For non-fibrous particles, size-selective air sampling instruments and direct reading instruments are used to measure mass concentrations of respirable particles. For fibrous particles, the approach is different. The measuring technique is based upon filter collection of “total dust” and counting of fibres under the optical microscope. In this case, the size selection is made by excluding from the count the “non-respirable” fibres with dimensions exceeding predetermined criteria.
Following the deposition of particles on the alveolar surfaces there starts the so-called alveolar clearance process. Chemotactic recruitment of macrophages and phagocytosis constitute its first phases. Several clearance pathways have been described: removal of dust-laden macrophages toward the ciliated airways, interaction with the epithelial cells and transfer of free particles through the alveolar membrane, phagocytosis by interstitial macrophages, sequestration into the interstitial area and transportation to the lymph nodes (Lauweryns and Baert 1977). Clearance pathways have specific kinetics. Not only the exposure regimen, but also the physicochemical characteristics of the deposited particles, trigger the activation of the different pathways responsible for the lung’s retention of such contaminants.
The notion of a retention pattern specific to each type of dust is rather new, but is now sufficiently established to be integrated into aetiopathogenesis schemes. For example, this author has found that after long term exposure to asbestos, fibres will accumulate in the lung if they are of the amphibole type, but will not if they are of the chrysotile type (Sébastien 1991). Short fibres have been shown to be cleared more rapidly than longer ones. Quartz is known to exhibit some lymph tropism and readily penetrates the lymphatic system. Modifying the surface chemistry of quartz particles has been shown to affect alveolar clearance (Hemenway et al. 1994; Dubois et al. 1988). Concomitant exposure to several dust types may also influence alveolar clearance (Davis, Jones and Miller 1991).
During alveolar clearance, dust particles may undergo some chemical and physical changes. Examples of theses changes include coating with ferruginous material, the leaching of some elemental constituents and the adsorption of some biological molecules.
Another notion recently derived from animal experiments is that of “lung overload” (Mermelstein et al. 1994). Rats heavily exposed by inhalation to a variety of insoluble dusts developed similar responses: chronic inflammation, increased numbers of particle-laden macrophages, increased numbers of particles in the interstitium, septal thickening, lipoproteinosis and fibrosis. These findings were not attributed to the reactivity of the dust tested (titanium dioxide, volcanic ash, fly ash, petroleum coke, polyvinyl chloride, toner, carbon black and diesel exhaust particulates), but to an excessive exposure of the lung. It is not known if lung overload must be considered in the case of human exposure to fibrogenic dusts.
Among the clearance pathways, the transfer towards the interstitium would be of particular importance for pneumoconioses. Clearance of particles having undergone sequestration into the interstitium is much less effective than clearance of particles engulfed by macrophages in the alveolar space and removed by ciliated airways (Vincent and Donaldson 1990). In humans, it was found that after long-term exposure to a variety of inorganic airborne contaminants, the storage was much greater in interstitial than alveolar macrophages (Sébastien et al. 1994). The view was also expressed that silica-induced pulmonary fibrosis involves the reaction of particles with interstitial rather than alveolar macrophages (Bowden, Hedgecock and Adamson 1989). Retention is responsible for the “dose”, a measure of the contact between the dust particles and their biological environment. A proper description of the dose would require that one know at each point in time the amount of dust stored in the several lung structures and cells, the physicochemical states of the particles (including the surface states), and the interactions between the particles and the pulmonary cells and fluids. Direct assessment of dose in humans is obviously an impossible task, even if methods were available to measure dust particles in several biological samples of pulmonary origin such as sputum, bronchoalveolar lavage fluid or tissue taken at biopsy or autopsy (Bignon, Sébastien and Bientz 1979). These methods were used for a variety of purposes: to provide information on retention mechanisms, to validate certain exposure information, to study the role of several dust types in pathogenic developments (e.g., amphiboles versus chrysotile exposure in asbestosis or quartz versus coal in CWP) and to assist in diagnosis.
But these direct measurements provide only a snapshot of retention at the time of sampling and do not allow the investigator to reconstitute dose data. New dosimetric models offer interesting perspectives in that regard (Katsnelson et al. 1994; Smith 1991; Vincent and Donaldson 1990). These models aim at assessing dose from exposure information by considering the probability of deposition and the kinetics of the different clearance pathways. Recently there was introduced into these models the interesting notion of “harmfulness delivery” (Vincent and Donaldson 1990). This notion takes into account the specific reactivity of the stored particles, each particle being considered as a source liberating some toxic entities into the pulmonary milieu. In the case of quartz particles for example, it could be hypothesized that some surface sites could be the source of active oxygen species. Models developed along such lines could also be refined to take into account the great interindividual variation generally observed with alveolar clearance. This was experimentally documented with asbestos, “high retainer animals” being at greater risk of developing asbestosis (Bégin and Sébastien 1989).
So far, these models were exclusively used by experimental pathologists. But they could also be useful to epidemiologists (Smith 1991). Most epidemiological studies looking at exposure response relationships relied on “cumulative exposure”, an exposure index obtained by integrating over time the estimated concentrations of airborne dust to which workers had been exposed (product of intensity and duration). The use of cumulative exposure has some limitations. Analyses based on this index implicitly assume that duration and intensity have equivalent effects on risk (Vacek and McDonald 1991).
Maybe the use of these sophisticated dosimetric models could provide some explanation for a common observation in the epidemiology of pneumoconioses: “the considerable between-work force differences” and this phenomenon was clearly observed for asbestosis (Becklake 1991) and for CWP (Attfield and Morring 1992). When relating the prevalence of the disease to the cumulative exposure, great differences—up to 50-fold—in risk were observed between some occupational groups. The geological origin of the coal (coal rank) provided partial explanation for CWP, mining deposits of high rank coal (a coal with high carbon content, like anthracite) yielding greater risk. The phenomenon remains to be explained in the case of asbestosis. Uncertainties on the proper exposure response curve have some bearings—at least theoretically—on the outcome, even at current exposure standards.
More generally, exposure metrics are essential in the process of risk assessment and the establishment of control limits. The use of the new dosimetric models may improve the process of risk assessment for pneumoconioses with the ultimate goal of increasing the degree of protection offered by control limits (Kriebel 1994).
Physicochemical Characteristics of Fibrogenic Dust Particles
A toxicity specific to each type of dust, related to the physicochemical characteristics of the particles (including the more subtle ones such as the surface characteristics), constitutes probably the most important notion to have emerged progressively during the last 20 years. In the very earliest stages of research, no differentiation were made among “mineral dusts”. Then generic categories were introduced: asbestos, coal, artificial inorganic fibres, phyllosilicates and silica. But this classification was found to be not precise enough to account for the variety in observed biological effects. Nowadays a mineralogical classification is used. For example, the several mineralogical types of asbestos are distinguished: serpentine chrysotile, amphibole amosite, amphibole crocidolite and amphibole tremolite. For silica, a distinction is generally made between quartz (by far the most prevalent), other crystalline polymorphs, and amorphous varieties. In the field of coal, high rank and low rank coals should be treated separately, since there is strong evidence that the risk of CWP and especially the risk of progressive massive fibrosis is much greater after exposure to dust produced in high rank coal mines.
But the mineralogical classification has also some limits. There is evidence, both experimental and epidemiological (taking into account “between-workforce differences”), that the intrinsic toxicity of a single mineralogical type of dust can be modulated by acting on the physicochemical characteristics of the particles. This raised the difficult question of the toxicological significance of each of the numerous parameters which can be used to describe a dust particle and a dust cloud. At the single particle level, several parameters can be considered: bulk chemistry, crystalline structure, shape, density, size, surface area, surface chemistry and surface charge. Dealing with dust clouds adds another level of complexity because of the distribution of these parameters (e.g., size distribution and the composition of mixed dust).
The size of the particles and their surface chemistry were the two parameters most studied to explain the modulation effect. As seen before, retention mechanisms are size related. But size may also modulate the toxicity in situ, as demonstrated by numerous animal and in vitro studies.
In the field of mineral fibres, the size was considered of so much importance that it constituted the basis of a pathogenesis theory. This theory attributed the toxicity of fibrous particles (natural and artificial) to the shape and size of the particles, leaving no role for the chemical composition. In dealing with fibres, size must be broken down into length and diameter. A two-dimensional matrix should be used to report size distributions, the useful ranges being 0.03 to 3.0mm for diameter and 0.3 to 300mm for length (Sébastien 1991). Integrating the results of the numerous studies, Lippman (1988) assigned a toxicity index to several cells of the matrix. There is a general tendency to believe that long and thin fibres are the most dangerous ones. Since the standards currently used in industrial hygiene are based upon the use of the optical microscope, they ignore the thinnest fibres. If assessing the specific toxicity of each cell within the matrix has some academic interest, its practical interest is limited by the fact that each type of fibre is associated with a specific size distribution that is relatively uniform. For compact particles, such as coal and silica, there is unclear evidence about a possible specific role for the different size sub-fractions of the particles deposited in the alveolar region of the lung.
More recent pathogenesis theories in the field of mineral dust imply active chemical sites (or functionalities) present at the surface of the particles. When the particle is “born” by separation from its parent material, some chemical bonds are broken in either a heterolytic or a homolytic way. What occurs during breaking and subsequent recombinations or reactions with ambient air molecules or biological molecules makes up the surface chemistry of the particles. Regarding quartz particles for example, several chemical functionalities of special interest have been described: siloxane bridges, silanol groups, partially ionized groups and silicon-based radicals.
These functionalities can initiate both acid-base and redox reactions. Only recently has attention been drawn to the latter (Dalal, Shi and Vallyathan 1990; Fubini et al. 1990; Pézerat et al. 1989; Kamp et al. 1992; Kennedy et al. 1989; Bronwyn, Razzaboni and Bolsaitis 1990). There is now good evidence that particles with surface-based radicals can produce reactive oxygen species, even in a cellular milieu. It is not certain if all the production of oxygen species should be attributed to the surface-based radicals. It is speculated that these sites may trigger the activation of lung cells (Hemenway et al. 1994). Other sites may be involved in the membranolytic activity of the cytotoxic particles with reactions such as ionic attraction, hydrogen bonding and hydrophobic bonding (Nolan et al. 1981; Heppleston 1991).
Following the recognition of surface chemistry as an important determinant of dust toxicity, several attempts were made to modify the natural surfaces of mineral dust particles to reduce their toxicity, as assessed in experimental models.
Adsorption of aluminium on quartz particles was found to reduce their fibrogenicity and to favour alveolar clearance (Dubois et al. 1988). Treatment with polyvinylpyridine-N-oxide (PVPNO) had also some prophylactic effect (Goldstein and Rendall 1987; Heppleston 1991). Several other modifying processes were used: grinding, thermal treatment, acid etching and adsorption of organic molecules (Wiessner et al. 1990). Freshly fractured quartz particles exhibited the highest surface activity (Kuhn and Demers 1992; Vallyathan et al. 1988). Interestingly enough, every departure from this “fundamental surface” led to a decrease in quartz toxicity (Sébastien 1990). The surface purity of several naturally occurring quartz varieties could be responsible for some observed differences in toxicity (Wallace et al. 1994). Some data support the idea that the amount of uncontaminated quartz surface is an important parameter (Kriegseis, Scharman and Serafin 1987).
The multiplicity of the parameters, together with their distribution in the dust cloud, yields a variety of possible ways to report air concentrations: mass concentration, number concentration, surface area concentration and concentration in various size categories. Thus, numerous indices of exposure can be constructed and the toxicological significance of each has to be assessed. The current standards in occupational hygiene reflect this multiplicity. For asbestos, the standards are based on the numerical concentration of fibrous particles in a certain geometrical size category. For silica and coal, the standards are based on the mass concentration of respirable particles. Some standards have also been developed for exposure to mixtures of particles containing quartz. No standard is based upon surface characteristics.
Biological Mechanisms Inducing the Fundamental Lesions
Pneumoconioses are interstitial fibrous lung diseases, the fibrosis being diffuse or nodular. The fibrotic reaction involves the activation of the lung fibroblast (Goldstein and Fine 1986) and the production and metabolism of the connective tissue components (collagen, elastin and glycosaminoglycans). It is considered to represent a late healing stage after lung injury (Niewoehner and Hoidal 1982). Even if several factors, essentially related to the characteristics of exposure, can modulate the pathological response, it is interesting to note that each type of pneumoconiosis is characterized by what could be called a fundamental lesion. The fibrosing alveolitis around the peripheral airways constitutes the fundamental lesion of asbestos exposure (Bégin et al. 1992). The silicotic nodule is the fundamental lesion of silicosis (Ziskind, Jones and Weil 1976). Simple CWP is composed of dust macules and nodules (Seaton 1983).
The pathogenesis of the pneumoconioses is generally presented as a cascade of events whose sequence runs as follows: alveolar macrophage alveolitis, signalling by inflammatory cell cytokines, oxidative damage, proliferation and activation of fibroblasts and the metabolism of collagen and elastin. Alveolar macrophage alveolitis is a characteristic reaction to retention of fibrosing mineral dust (Rom 1991). The alveolitis is defined by increased numbers of activated alveolar macrophages releasing excessive quantities of mediators including oxidants, chemotaxins, fibroblast growth factors and protease. Chemotaxins attract neutrophils and, together with macrophages, may release oxidants capable of injuring alveolar epithelial cells. Fibroblast growth factors gain access to the interstitium, where they signal fibroblasts to replicate and increase the production of collagen.
The cascade starts at the first encounter of particles deposited in the alveoli. With asbestos for example, the initial lung injury occurs almost immediately after exposure at the alveolar duct bifurcations. After only 1 hour of exposure in animal experiments, there is active uptake of fibres by type I epithelial cells (Brody et al. 1981). Within 48 hours, increased numbers of alveolar macrophages accumulate at sites of deposition. With chronic exposure, this process may lead to peribronchiolar fibrosing alveolitis.
The exact mechanism by which deposited particles produce primary biochemical injury to the alveolar lining, a specific cell, or any of its organelles, is unknown. It may be that extremely rapid and complex biochemical reactions result in free radical formation, lipid peroxidation, or a depletion in some species of vital cell protectant molecule. It has been shown that mineral particles can act as catalytic substrates for hydroxyl and superoxide radical generation (Guilianelli et al. 1993).
At the cellular level, there is slightly more information. After deposition at the alveolar level, the very thin epithelial type I cell is readily damaged (Adamson, Young and Bowden 1988). Macrophages and other inflammatory cells are attracted to the damage site and the inflammatory response is amplified by the release of arachidonic acid metabolites such as prostaglandins and leukotrienes together with exposure of the basement membrane (Holtzman 1991; Kuhn et al. 1990; Engelen et al. 1989). At this stage of primary damage, the lung architecture becomes disorganized, showing an interstitial oedema.
During the chronic inflammatory process, both the surface of the dust particles and the activated inflammatory cells release increased amounts of reactive oxygen species in the lower respiratory tract. The oxidative stress in the lung has some detectable effects on the antioxidant defense system (Heffner and Repine 1989), with expression of antioxidant enzymes like superoxide dismutase, glutathione peroxidases and catalase (Engelen et al. 1990). These factors are located in the lung tissue, the interstitial fluid and the circulating erythrocytes. The profiles of antioxidant enzymes may depend on the type of fibrogenic dust (Janssen et al. 1992). Free radicals are known mediators of tissue injury and disease (Kehrer 1993).
Interstitial fibrosis does result from a repair process. There are numerous theories to explain how the repair process takes place. The macrophage/fibroblast interaction has received the greatest attention. Activated macrophages secrete a network of proinflammatory fibrogenic cytokines: TNF, IL-1, transforming growth factor and platelet-derived growth factor. They also produce fibronectin, a cell surface glycoprotein which acts as a chemical attractant and, under some conditions, as a growth stimulant for mesenchymal cells. Some authors consider that some factors are more important than others. For example, special importance was ascribed to TNF in the pathogenesis of silicosis. In experimental animals, it was shown that collagen deposition after silica instillation in mice was almost completely prevented by anti-TNF antibody (Piguet et al. 1990). The release of platelet-derived growth factor and transforming growth factor was presented as playing an important role in the pathogenesis of asbestosis (Brody 1993).
Unfortunately, many of the macrophage/fibroblast theories tend to ignore the potential balance between the fibrogenic cytokines and their inhibitors (Kelley 1990). In fact, the resulting imbalance between oxidizing and antioxidizing agents, proteases and antiproteases, the arachidonic acid metabolites, elastases and collagenases, as well as the imbalances between the various cytokines and growth factors, would determine the abnormal remodelling of the interstitium component towards the several forms of pneumoconioses (Porcher et al. 1993). In pneumoconioses, the balance is clearly directed towards an overwhelming effect of the damaging cytokine activities.
Because type I cells are incapable of division, after the primary insult, the epithelial barrier is replaced with type II cells (Lesur et al. 1992). There is some indication that if this epithelial repair process is successful and that the regenerating type II cells are not damaged further, the fibrogenesis is not likely to proceed. Under some conditions, the repair by the type II cell is taken to excess, resulting in alveolar proteinosis. This process was clearly demonstrated after silica exposure (Heppleston 1991). To what extent the alterations in epithelial cells influence the fibroblasts is uncertain. Thus, it would seem that fibrogenesis is initiated in areas of extensive epithelial damage, as fibroblasts replicate, then differentiate and produce more collagen, fibronectin and other components of the extracelluar matrix.
There is abundant literature on the biochemistry of the several types of collagen formed in pneumoconioses (Richards, Masek and Brown 1991). The metabolism of such collagen and its stability in the lung are important elements of the fibrogenesis process. The same probably holds for the other components of the damaged connective tissue. The metabolism of collagen and elastin is of particular interest in the healing phase since these proteins are so important to lung structure and function. It has been very nicely shown that alterations in the synthesis of these proteins might determine whether emphysema or fibrosis evolves after lung injury (Niewoehner and Hoidal 1982). In the disease state, mechanisms such as an increase in transglutaminase activity could favour the formation of stable protein masses. In some CWP fibrotic lesions, the protein components account for one-third of the lesion, the rest being dust and calcium phosphate.
Considering only collagen metabolism, several stages of fibrosis are possible, some of which are potentially reversible while others are progressive. There is experimental evidence that unless a critical exposure is exceeded, the early lesions can regress and irreversible fibrosis is an unlikely outcome. In asbestosis for example, several types of lung reactions were described (Bégin, Cantin and Massé 1989): a transient inflammatory reaction without lesion, a low retention reaction with fibrotic scar limited to the distal airways, a high inflammatory reaction sustained by the continuous exposure and the weak clearance of the longest fibres.
It can be concluded from these studies that exposure to fibrotic dust particles is able to trigger several complex biochemical and cellular pathways involved in lung injury and repair. Exposure regimen, physicochemical characteristics of the dust particles, and possibly individual susceptibility factors seem to be the determinants of the fine balance among the several pathways. Physicochemical characteristics will determine the type of the ultimate fundamental lesion. Exposure regimen seems to determine the time course of events. There is some indication that sufficiently low exposure regimens can in most cases limit the lung reaction to non-progressive lesions with no disability or impairment.
Medical surveillance and screening always have been part of the strategies for the prevention of pneumoconioses. In that context, the possibility of detecting some early lesions is advantageous. Increased knowledge of pathogenesis paved the way to the development of several biomarkers (Borm 1994) and to the refinement and use of “non-classical” pulmonary investigation techniques such as the measurement of the clearance rate of deposited 99 technetium diethylenetriamine-penta-acetate (99 Tc-DTPA) to assess pulmonary epithelial integrity (O’Brodovich and Coates 1987), and quantitative gallium-67 lung scan to assess inflammatory activity (Bisson, Lamoureux and Bégin 1987).
Several biomarkers were considered in the field of pneumoconioses: sputum macrophages, serum growth factors, serum type III procollagen peptide, red blood cell antioxidants, fibronectin, leucocyte elastase, neutral metalloendopeptidase and elastin peptides in plasma, volatile hydrocarbons in exhaled air and TNF release by peripheral blood monocytes. Biomarkers are conceptually quite interesting, but many more studies are necessary to assess their significance precisely. This validation effort will be quite demanding, since it will require investigators to conduct prospective epidemiological studies. Such an effort was carried out recently for TNF release by peripheral blood monocytes in CWP. TNF was found to be an interesting marker of CWP progression (Borm 1994). Besides the scientific aspects of the significance of biomarkers in the pathogenesis of pneumoconioses, other issues related to the use of biomarkers must be examined carefully (Schulte 1993), namely, opportunities for prevention, impact on occupational medicine and ethical and legal problems.
Progression and Complication of Pneumoconioses
In the early decades of this century, pneumoconiosis was regarded as a disease that disabled the young and killed prematurely. In industrialized countries, it is now generally regarded as no more than a radiological abnormality, without impairment or disability (Sadoul 1983). However, two observations should be set against this optimistic statement. First, even if under limited exposure, pneumoconiosis remains a relatively silent and asymptomatic disease, it should be known that the disease may progress towards more severe and disabling forms. Factors affecting this progression are definitely important to consider as part of the aetiopathogenesis of the condition. Secondly, there is now evidence that some pneumoconioses can affect general health outcome and can be a contributing factor for lung cancer.
The chronic and progressive nature of asbestosis has been documented from the initial subclinical lesion to clinical asbestosis (Bégin, Cantin and Massé 1989). Modern pulmonary investigation techniques (BAL, CT scan, gallium-67 lung uptake) revealed that inflammation and injury was continuous from the time of exposure, through the latent or subclinical phase, to the development of the clinical disease. It has been reported (Bégin et al. 1985) that 75% of subjects who initially had a positive gallium-67 scan but did not have clinical asbestosis at that time, did progress to “full-blown” clinical asbestosis over a four-year period. In both humans and experimental animals, asbestosis may progress after disease recognition and exposure cessation. It is highly probable that exposure history prior to recognition is an important determinant of progression. Some experimental data support the notion of non-progressive asbestosis associated with light induction exposure and exposure cessation at recognition (Sébastien, Dufresne and Bégin 1994). Assuming that the same notion applies to humans, it would be of the first importance to establish precisely the metrics of “light induction exposure”. In spite of all the efforts at screening working populations exposed to asbestos, this information is still lacking.
It is well-known that asbestos exposure can yield to an excessive risk of lung cancer. Even if it is admitted that asbestos is a carcinogen per se, it has long been debated whether the risk of lung cancer among asbestos workers was related to the exposure to asbestos or to the lung fibrosis (Hughes and Weil 1991). This issue is not resolved yet.
Owing to continuous improvement of working conditions in modern mining facilities, nowadays, CWP is a disease affecting essentially retired miners. If simple CWP is a condition without symptoms and without demonstrable effect on lung function, progressive massive fibrosis (PMF) is a much more severe condition, with major structural alterations of the lung, deficits of lung function and reduced life expectancy. Many studies have aimed at identifying the determinants of progression towards PMF (heavy retention of dust in the lung, coal rank, mycobacterial infection or immunological stimulation). A unifying theory was proposed (Vanhee et al. 1994), based upon a continuous and severe alveolar inflammation with activation of the alveolar macrophages and substantial production of reactive oxygen species, chemotactic factors and fibronectin. Other complications of CWP include mycobacterial infection, Caplan’s syndrome and scleroderma. There is no evidence of elevated risk of lung cancer among coal miners.
The chronic form of silicosis follows exposure, measured in decades rather than years, to respirable dust containing generally less than 30% quartz. But in case of uncontrolled exposure to quartz-rich dust (historical exposures with sand blasting, for example), acute and accelerated forms can be found after only several months. Cases of acute and accelerated disease are particularly at risk of complication by tuberculosis (Ziskind, Jones and Weil 1976). Progression may also occur, with the development of large lesions that obliterate lung structure, called either complicated silicosis or PMF.
A few studies examined the progression of silicosis in relation to exposure and yielded diverging results about the relationships between progression and exposure, before and after onset (Hessel et al. 1988). Recently, Infante-Rivard et al. (1991) studied the prognostic factors influencing the survival of compensated silicotic patients. Patients with small opacities alone on their chest radiograph and who did not have dyspnoea, expectoration or abnormal breath sounds had a survival similar to that of the referents. Other patients had a poorer survival. Finally, one should mention the recent concern about silica, silicosis and lung cancer. There is some evidence for and against the proposition that silica per se is carcinogenic (Agius 1992). Silica may synergize potent environmental carcinogens, such as those in tobacco smoke, through a relatively weak promoting effect on carcinogenesis or by impairing their clearance. Moreover, the disease process associated with or leading to silicosis might carry an increased risk of lung cancer.
Nowadays, progression and complication of pneumoconioses could be considered as a key issue for medical management. The use of classical pulmonary investigation techniques has been refined for early recognition of the disease (Bégin et al. 1992), at a stage where pneumoconiosis is limited to its radiological manifestation, without impairment or disability. In the near future, it is probable that a battery of biomarkers will be available to document even earlier stages of the disease. The question of whether a worker diagnosed with pneumoconiosis—or documented to be in its earlier stages—should be allowed to continue with his or her job has puzzled occupational health decision makers for some time. It is a rather difficult question which entails ethical, social and scientific considerations. If an overwhelming scientific literature is available on the induction of pneumoconiosis, the information on progression usable by decision makers is rather sparse and somewhat confusing. A few attempts were made to study the roles of variables such as exposure history, dust retention and medical condition at onset. The relationships between all these variables do complicate the issue. Recommendations are made for health screening and surveillance of workers exposed to mineral dust (Wagner 1996). Programmes are already—or will be—put in place accordingly. Such programmes would definitely benefit from better scientific knowledge on progression, and especially on the relation between exposure and retention characteristics.
Discussion
The information brought by many scientific disciplines to bear upon the aetiopathogenesis of the pneumoconioses is overwhelming. The major difficulty now is to reassemble the scattered elements of the puzzle into unifying mechanistic pathways leading to the fundamental lesions of the pneumoconioses. Without this necessary integration, we would be left with the contrast between a few fundamental lesions, and very numerous biochemical and cellular reactions.
Our knowledge of aetiopathogenesis has so far influenced the practices of occupational hygiene only to a limited extent, in spite of the strong intention of hygienists to operate according to standards having some biological significance. Two main notions were incorporated in their practices: the size selection of respirable dust particles and the dust type dependence of toxicity. The latter yielded some limits specific to each type of dust. The quantitative risk assessment, a necessary step in defining exposure limits, constitutes a complicated exercise for several reasons, such as the variety of possible exposure indices, poor information on past exposure, the difficulty one has with epidemiological models in dealing with multiple indices of exposure and the difficulty in estimating dose from exposure information. The current exposure limits, embodying sometimes considerable uncertainty, are probably low enough to offer good protection. The between-workforce differences observed in exposure-response relationships however, reflect our incomplete control of the phenomenon.
The impact of newer understanding of the cascade of events in the pathogenesis of the pneumoconioses has not modified the traditional approach to workers’ surveillance, but has significantly helped physicians in their capacity of recognizing the disease (pneumoconiosis) early, at a time when the disease has had only a limited impact on lung function. It is indeed subjects at the early stage of disease that should be recognized and withdrawn from further significant exposure if prevention of disability is to be achieved by medical surveillance.
Silicosis
Silicosis is a fibrotic disease of the lungs caused by the inhalation, retention and pulmonary reaction to crystalline silica. Despite knowledge of the cause of this disorder—respiratory exposures to silica containing dusts—this serious and potentially fatal occupational lung disease remains prevalent throughout the world. Silica, or silicon dioxide, is the predominant component of the earth’s crust. Occupational exposure to silica particles of respirable size (aerodynamic diameter of 0.5 to 5μm) is associated with mining, quarrying, drilling, tunnelling and abrasive blasting with quartz containing materials (sandblasting). Silica exposure also poses a hazard to stonecutters, and pottery, foundry, ground silica and refractory workers. Because crystalline silica exposure is so widespread and silica sand is an inexpensive and versatile component of many manufacturing processes, millions of workers throughout the world are at risk of the disease. The true prevalence of the disease is unknown.
Definition
Silicosis is an occupational lung disease attributable to the inhalation of silicon dioxide, commonly known as silica, in crystalline forms, usually as quartz, but also as other important crystalline forms of silica, for example, cristobalite and tridymite. These forms are also called “free silica” to distinguish them from the silicates. The silica content in different rock formations, such as sandstone, granite and slate, varies from 20 to nearly 100%.
Workers in High-Risk Occupations and Industries
Although silicosis is an ancient disease, new cases are still reported in both the developed and developing world. In the early part of this century, silicosis was a major cause of morbidity and mortality. Contemporary workers are still exposed to silica dust in a variety of occupations—and when new technology lacks adequate dust control, exposures may be to more hazardous dust levels and particles than in non-mechanized work settings. Whenever the earth’s crust is disturbed and silica-containing rock or sand is used or processed, there are potential respiratory risks for workers. Reports continue of silicosis from industries and work settings not previously recognized to be at risk, reflecting the nearly ubiquitous presence of silica. Indeed, due to the latency and chronicity of this disorder, including the development and progression of silicosis after exposure has ceased, some workers with current exposures may not manifest disease until the next century. In many countries throughout the world, mining, quarrying, tunnelling, abrasive blasting and foundry work continue to present major risks for silica exposure, and epidemics of silicosis continue to occur, even in developed nations.
Forms of Silicosis—Exposure History and Clinicopathologic Descriptions
Chronic, accelerated and acute forms of silicosis are commonly described. These clinical and pathologic expressions of the disease reflect differing exposure intensities, latency periods and natural histories. The chronic or classic form usually follows one or more decades of exposure to respirable dust containing quartz, and this may progress to progressive massive fibrosis (PMF). The accelerated form follows shorter and heavier exposures and progresses more rapidly. The acute form may occur after short-term, intense exposures to high levels of respirable dust with high silica content for periods that may be measured in months rather than years.
Chronic (or classic) silicosis may be asymptomatic or result in insidiously progressive exertional dyspnoea or cough (often mistakenly attributed to the ageing process). It presents as a radiographic abnormality with small (<10 mm), rounded opacities predominantly in the upper lobes. A history of 15 years or more since onset of exposure is common. The pathologic hallmark of the chronic form is the silicotic nodule. The lesion is characterized by a cell-free central area of concentrically arranged, whorled hyalinized collagen fibers, surrounded by cellular connective tissue with reticulin fibers. Chronic silicosis may progress to PMF (sometimes referred to as complicated silicosis), even after exposure to silica-containing dust has ceased.
Progressive massive fibrosis is more likely to present with exertional dyspnoea. This form of disease is characterized by nodular opacities greater than 1 cm on chest radiograph and commonly will involve reduced carbon monoxide diffusing capacity, reduced arterial oxygen tension at rest or with exercise, and marked restriction on spirometry or lung volume measurement. Distortion of the bronchial tree may also lead to airway obstruction and productive cough. Recurrent bacterial infection not unlike that seen in bronchiectasis may occur. Weight loss and cavitation of the large opacities should prompt concern for tuberculosis or other mycobacterial infection. Pneumothorax may be a life-threatening complication, since the fibrotic lung may be difficult to re-expand. Hypoxaemic respiratory failure with cor pulmonale is a common terminal event.
Accelerated silicosis may appear after more intense exposures of shorter (5 to 10 years) duration. Symptoms, radiographic findings and physiological measurements are similar to those seen in the chronic form. Deterioration in lung function is more rapid, and many workers with accelerated disease may develop mycobacterial infection. Auto-immune disease, including scleroderma or systemic sclerosis, is seen with silicosis, often of the accelerated type. The progression of radiographic abnormalities and functional impairment can be very rapid when auto-immune disease is associated with silicosis.
Acute silicosis may develop within a few months to 2 years of massive silica exposure. Dramatic dyspnoea, weakness, and weight loss are often presenting symptoms. The radiographic findings of diffuse alveolar filling differ from those in the more chronic forms of silicosis. Histologic findings similar to pulmonary alveolar proteinosis have been described, and extrapulmonary (renal and hepatic) abnormalities are occasionally reported. Rapid progression to severe hypoxaemic ventilatory failure is the usual course.
Tuberculosis may complicate all forms of silicosis, but people with acute and accelerated disease may be at highest risk. Silica exposure alone, even without silicosis may also predispose to this infection. M. tuberculosis is the usual organism, but atypical mycobacteria are also seen.
Even in the absence of radiographic silicosis, silica-exposed workers may also have other diseases associated with occupational dust exposure, such as chronic bronchitis and the associated emphysema. These abnormalities are associated with many occupational mineral dust exposures, including dusts containing silica.
Pathogenesis and the Association with Tuberculosis
The precise pathogenesis of silicosis is uncertain, but an abundance of evidence implicates the interaction between the pulmonary alveolar macrophage and silica particles deposited in the lung. Surface properties of the silica particle appear to promote macrophage activation. These cells then release chemotactic factors and inflammatory mediators that result in a further cellular response by polymorphonuclear leukocytes, lymphocytes and additional macrophages. Fibroblast-stimulating factors are released that promote hyalinization and collagen deposition. The resulting pathologic silicotic lesion is the hyaline nodule, containing a central acellular zone with free silica surrounded by whorls of collagen and fibroblasts, and an active peripheral zone composed of macrophages, fibroblasts, plasma cells, and additional free silica as shown in figure 1.
Figure 1. Typical silicotic nodule, microscopic section. Courtesy of Dr. V. Vallyathan.
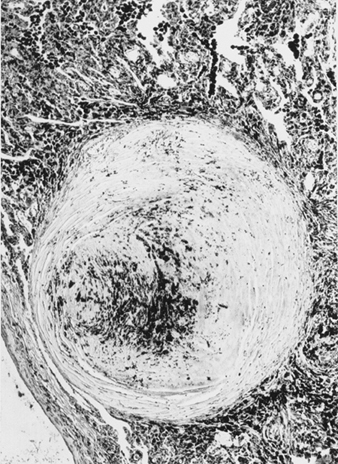
The precise properties of silica particles that evoke the pulmonary response described above are not known, but surface characteristics may be important. The nature and the extent of the biological response are in general related to the intensity of the exposure; however, there is growing evidence that freshly fractured silica may be more toxic than aged dust containing silica, an effect perhaps related to reactive radical groups on the cleavage planes of freshly fractured silica. This may offer a pathogenic explanation for the observation of cases of advanced disease in both sandblasters and rock drillers where exposures to recently fractured silica are particularly intense.
The initiating toxic insult may occur with minimal immunological reaction; however, a sustained immunological response to the insult may be important in some of the chronic manifestations of silicosis. For example, antinuclear antibodies may occur in accelerated silicosis and scleroderma, as well as other collagen diseases in workers who have been exposed to silica. The susceptibility of silicotic workers to infections, such as tuberculosis and Nocardia asteroides, is likely related to the toxic effect of silica on pulmonary macrophages.
The link between silicosis and tuberculosis has been recognized for nearly a century. Active tuberculosis in silicotic workers may exceed 20% when community prevalence of tuberculosis is high. Again, people with acute silicosis appear to be at considerably higher risk.
Clinical Picture of Silicosis
The primary symptom is usually dyspnoea, first noted with activity or exercise and later at rest as the pulmonary reserve of the lung is lost. However, in the absence of other respiratory disease, shortness of breath may be absent and the presentation may be an asymptomatic worker with an abnormal chest radiograph. The radiograph may at times show quite advanced disease with only minimal symptoms. The appearance or progression of dyspnoea may herald the development of complications including tuberculosis, airways obstruction or PMF. Cough is often present secondary to chronic bronchitis from occupational dust exposure, tobacco use, or both. Cough may at times also be attributed to pressure from large masses of silicotic lymph nodes on the trachea or mainstem bronchi.
Other chest symptoms are less common than dyspnoea and cough. Haemoptysis is rare and should raise concern for complicating disorders. Wheeze and chest tightness may occur usually as part of associated obstructive airways disease or bronchitis. Chest pain and finger clubbing are not features of silicosis. Systemic symptoms, such as fever and weight loss, suggest complicating infection or neoplastic disease. Advanced forms of silicosis are associated with progressive respiratory failure with or without cor pulmonale. Few physical signs may be noted unless complications are present.
Radiographic Patterns and Functional Pulmonary Abnormalities
The earliest radiographic signs of uncomplicated silicosis are generally small rounded opacities. These can be described by the ILO International Classification of Radiographs of Pneumoconioses by size, shape and profusion category. In silicosis, “q” and “r” type opacities dominate. Other patterns including linear or irregular shadows have also been described. The opacities seen on the radiograph represent the summation of pathologic silicotic nodules. They are usually found predominantly in the upper zones and may later progress to involve other zones. Hilar lymphadenopathy is also noted sometimes in advance of nodular parenchymal shadows. Egg shell calcification is strongly suggestive of silicosis, although this feature is seen infrequently. PMF is characterized by the formation of large opacities. These large lesions can be described by size using the ILO classification as categories A, B or C. Large opacities or PMF lesions tend to contract, usually to the upper lobes, leaving areas of compensatory emphysema at their margins and often in the lung bases. As a result, previously evident small rounded opacities may disappear at times or be less prominent. Pleural abnormalities may occur but are not a frequent radiographic feature in silicosis. Large opacities may also pose concern regarding neoplasm and radiographic distinction in the absence of old films may be difficult. All lesions that cavitate or change rapidly should be evaluated for active tuberculosis. Acute silicosis may present with a radiologic alveolar filling pattern with rapid development of PMF or complicated mass lesions. See figures 2 and 3.
Figure 2. Chest radiograph, acute silico-proteinosis in a surface coal mine driller. Courtesy of Dr. NL Lapp and Dr. DE Banks.
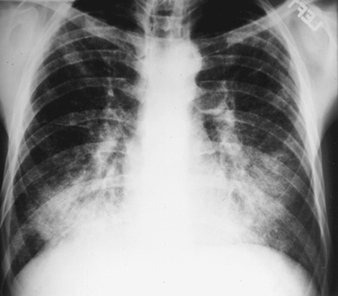
Figure 3. Chest radiograph, complicated silicosis demonstrating progressive massive fibrosis.
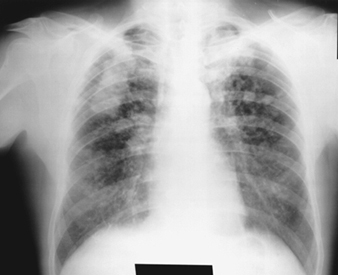
Pulmonary function tests, such as spirometry and diffusing capacity, are helpful for the clinical evaluation of people with suspected silicosis. Spirometry may also be of value in early recognition of the health effects from occupational dust exposures, as it may detect physiologic abnormalities that may precede radiologic changes. No solely characteristic pattern of ventilatory impairment is present in silicosis. Spirometry may be normal, or when abnormal, the tracings may show obstruction, restriction or a mixed pattern. Obstruction may indeed be the more common finding. These changes tend to be more marked with advanced radiologic categories. However, poor correlation exists between radiographic abnormalities and ventilatory impairment. In acute and accelerated silicosis, functional changes are more marked and progression is more rapid. In acute silicosis, radiologic progression is accompanied by increasing ventilatory impairment and gas exchange abnormalities, which leads to respiratory failure and eventually to death from intractable hypoxaemia.
Complications and Special Diagnostic Issues
With a history of exposure and a characteristic radiograph, the diagnosis of silicosis is generally not difficult to establish. Challenges arise only when the radiologic features are unusual or the history of exposure is not recognized. Lung biopsy is rarely required to establish the diagnosis. However, tissue samples are helpful in some clinical settings when complications are present or the differential diagnosis includes tuberculosis, neoplasm or PMF. Biopsy material should be sent for culture, and in research settings, dust analysis may be a useful additional measure. When tissue is required, open lung biopsy is generally necessary for adequate material for examination.
Vigilance for infectious complications, especially tuberculosis, cannot be overemphasized, and symptoms of change in cough or hemoptysis, and fever or weight loss should trigger a work-up to exclude this treatable problem.
Substantial concern and interest about the relationship between silica exposure, silicosis and cancer of the lung continues to stimulate debate and further research. In October of 1996, a committere of The International Agency for Research on Cancer (IARC) classified crystalline silica as a Group I carcinogen, reaching this conclusion based on “sufficient evidence of carcinogenicity in humans”. Uncertainty over the pathogenic mechanisms for the development of lung cancer in silica-exposed populations exists, and the possible relationship between silicosis (or lung fibrosis) and cancer in exposed workers continues to be studied. Regardless of the mechanism that may be responsible for neoplastic events, the known association between silica exposures and silicosis dictates controlling and reducing exposures to workers at risk for this disease.
Prevention of Silicosis
Prevention remains the cornerstone of eliminating this occupational lung disease. The use of improved ventilation and local exhaust, process enclosure, wet techniques, personal protection including the proper selection of respirators, and where possible, industrial substitution of agents less hazardous than silica all reduce exposure. The education of workers and employers regarding the hazards of silica dust exposure and measures to control exposure is also important.
If silicosis is recognized in a worker, removal from continuing exposure is advisable. Unfortunately, the disease may progress even without further silica exposure. Additionally, finding a case of silicosis, especially the acute or accelerated form, should prompt a workplace evaluation to protect other workers also at risk.
Screening and Surveillance
Silica and other mineral-dust exposed workers should have periodic screening for adverse health effects as a supplement to, but not a substitute for, dust exposure control. Such screening commonly includes evaluations for respiratory symptoms, lung function abnormalities, and neoplastic disease. Evaluations for tuberculosis infection should also be performed. In addition to individual worker screening, data from groups of workers should be collected for surveillance and prevention activities. Guidance for these types of studies is included in the list of suggested readings.
Therapy, Management of Complications and Control of Silicosis
When prevention has been unsuccessful and silicosis has developed, therapy is directed largely at complications of the disease. Therapeutic measures are similar to those commonly used in the management of airway obstruction, infection, pneumothorax, hypoxaemia, and respiratory failure complicating other pulmonary disease. Historically, the inhalation of aerosolized aluminium has been unsuccessful as a specific therapy for silicosis. Polyvinyl pyridine-N-oxide, a polymer that has protected experiment animals, is not available for use in humans. Recent laboratory work with tetrandrine has shown in vivo reduction in fibrosis and collagen synthesis in silica exposed animals treated with this drug. However, strong evidence of human efficacy is currently lacking, and there are concerns about the potential toxicity, including the mutagenicity, of this drug. Because of the high prevalence of disease in some countries, investigations of combinations of drugs and other interventions continue. Currently, no successful approach has emerged, and the search for a specific therapy for silicosis to date has been unrewarding.
Further exposure is undesirable, and advice on leaving or changing the current job should be given with information about past and present exposure conditions.
In the medical management of silicosis, vigilance for complicating infection, especially tuberculosis, is critical. The use of BCG in the tuberculin-negative silicotic patient is not recommended, but the use of preventive isoniazid (INH) therapy in the tuberculin-positive silicotic subject is advised in countries where the prevalence of tuberculosis is low. The diagnosis of active tuberculosis infection in patients with silicosis can be difficult. Clinical symptoms of weight loss, fever, sweats and malaise should prompt radiographic evaluation and sputum acid-fast bacilli strains and cultures. Radiographic changes, including enlargement or cavitation in conglomerate lesions or nodular opacities, are of particular concern. Bacteriological studies on expectorated sputum may not always be reliable in silicotuberculosis. Fiberoptic bronchoscopy for additional specimens for culture and study may often be helpful in establishing a diagnosis of active disease. The use of multidrug therapy for suspected active disease in silicotics is justified at a lower level of suspicion than in the non-silicotic subject, due to the difficulty in firmly establishing evidence for active infection. Rifampin therapy appears to have enhanced the success rate of treatment of silicosis complicated by tuberculosis, and in some recent studies response to short-term therapy was comparable in cases of silicotuberculosis to that in matched cases of primary tuberculosis.
Ventilatory support for respiratory failure is indicated when precipitated by a treatable complication. Pneumothorax, spontaneous and ventilator-related, is usually treated by chest tube insertion. Bronchopleural fistula may develop, and surgical consultation and management should be considered.
Acute silicosis may rapidly progress to respiratory failure. When this disease resembles pulmonary alveolar proteinosis and severe hypoxaemia is present, aggressive therapy has included massive whole-lung lavage with the patient under general anaesthesia in an attempt to improve gas exchange and remove alveolar debris. Although appealing in concept, the efficacy of whole lung lavage has not been established. Glucocorticoid therapy has also been used for acute silicosis; however, it is still of unproven benefit.
Some young patients with end-stage silicosis may be considered candidates for lung or heart-lung transplantation by centres experienced with this expensive and high-risk procedure. Early referral and evaluation for this intervention may be offered to selected patients.
The discussion of an aggressive and high-technology therapeutic intervention such as transplantation serves dramatically to underscore the serious and potentially fatal nature of silicosis, as well as to emphasize the crucial role for primary prevention. The control of silicosis ultimately depends upon the reduction and control of workplace dust exposures. This is accomplished by rigorous and conscientious application of fundamental occupational hygiene and engineering principles, with a commitment to the preservation of worker health.
More...
Coal Workers' Lung Diseases
Coal miners are subject to a number of lung diseases and disorders arising from their exposure to coal mine dust. These include pneumoconiosis, chronic bronchitis and obstructive lung disease. The occurrence and severity of disease depends on the intensity and duration of dust exposure. The specific composition of the coal mine dust also has a bearing on some health outcomes.
In the developed countries, where high prevalences of lung disease existed in the past, reductions in dust levels brought about by regulation have led to substantial drops in disease prevalence since the 1970s. In addition, major reductions in the mining work force in most of those countries over recent decades, partly brought about by changes in technology and resulting improvements in productivity, will result in further reductions in overall disease levels. Miners in other countries, where coal mining is a more recent phenomenon and dust controls are less aggressive, have not been so fortunate. This problem is exacerbated by the high cost of modern mining technology, forcing the employment of large numbers of workers, many of whom are at high risk of disease development.
In the following text, each disease or disorder is considered in turn. Those specific to coal mining, such as coal workers’ pneumoconiosis are described in detail; the description of others, such as obstructive lung disease, is restricted to those aspects that relate to coal miners and dust exposure.
Coal Workers’ Pneumoconiosis
Coal workers’ pneumoconiosis (CWP) is the disease most commonly associated with coal mining. It is not a fast-developing disease, usually taking at least ten years to be manifested, and often much longer when exposures are low. In its initial stages it is an indicator of excessive lung dust retention, and may be associated with few symptoms or signs in itself. However, as it advances, it puts the miner at increasing risk of development of the much more serious progressive massive fibrosis (PMF).
Pathology
The classic lesion of CWP is the coal macule, a collection of dust and dust-laden macrophages around the periphery of the respiratory bronchioles. The macules contain minimal collagen and are thus usually not palpable. They are about 1 to 5 mm in size, and are frequently accompanied by an enlargement of the adjacent air spaces, termed focal emphysema. Though often very numerous, they are not usually evident on a chest radiograph.
Another lesion associated with CWP is the coal nodule. These larger lesions are palpable and contain a mixture of dust-laden macrophages, collagen and reticulin. The presence of coal nodules, with or without silicotic nodules (see below), indicates lung fibrosis, and is largely responsible for the opacities seen on chest radiographs. Macronodules (7 to 20 mm) in size may coalesce to form progressive massive fibrosis (see below), or PMF may develop from a single macronodule.
Silicotic nodules (described under silicosis) have been found in a significant minority of underground coal miners. For most, the cause may rest simply with the silica present in the coal dust, although exposure to pure silica in some jobs is certainly an important factor (e.g., among surface drillers, underground motormen and roof bolters).
Radiography
The most useful indicator of CWP in miners during life is obtained using the routine chest radiograph. Dust deposits and the nodular tissue reactions attenuate the x-ray beam and result in opacities on the film. The profusion of these opacities can be assessed systematically by using a standardized method of radiograph description such as that disseminated by the ILO and described else in this chapter. In this method, individual posterior-anterior films are compared to standard radiographs showing increasing profusion of small opacities, and the film classified into one of four major categories (0, 1, 2, 3) based on its similarity to the standard. A secondary classification is also made, depending on the reader’s assessment of the film’s similarity to adjacent ILO categories. Other aspects of the opacities, such as size, shape and region of occurrence in the lung are also noted. Some countries, such as China and Japan, have developed similar systems for systematic radiograph description or interpretation that are particularly suited to their own needs.
Traditionally, small rounded types of opacity have been associated with coal mining. However, more recent data indicate that irregular types can also result from exposure to coal mine dust. The opacities of CWP and silicosis are often indistinguishable on the radiograph. However, there is some evidence that larger sized opacities (type r) more often indicate silicosis.
It is important to note that a substantial amount of pathologic abnormality related to pneumoconiosis may be present in the lung before it can be detected on the routine chest x ray. This is particularly true for macular deposition, but it becomes progressively less true with greater profusion and size of nodules. Concomitant emphysema may also reduce the visibility of lesions on the chest x ray. Computerized tomography (CT)—particularly high-resolution computerized tomography (HRCT)—may permit visualization of abnormalities not clearly evident on routine chest x rays, although CT is not necessary for routine clinical diagnosis of miners’ lung diseases and is not indicated for medical surveillance of miners.
Clinical aspects
The development of CWP, although a marker of excessive lung dust retention, in itself is often unaccompanied by any overt clinical signs. This should not, however, be taken to imply that the inhalation of coal mine dust is without risk, for it is now well known that other lung diseases can arise from dust exposure. Pulmonary hypertension is more often noted in miners who develop airflow obstruction in association with CWP. Moreover, once CWP has developed, it usually progresses unless dust exposure ceases, and may progress thereafter. It also puts the miner at greatly increased risk of development of the clinically ominous PMF, with the likelihood of subsequent impairment, disability and premature mortality.
Disease mechanisms
Development of the earliest change of CWP, the dust macule, represents the effects of dust deposition and accumulation. The subsequent stage, that is, the development of nodules, results from the lung’s inflammatory and fibrotic reaction to the dust. In this, the roles of silica and non-silica dust have long been debated. On the one hand, silica dust is known to be considerably more toxic than coal dust. Yet, on the other hand, epidemiological studies have shown no strong evidence implicating silica exposure in CWP prevalence or incidence. Indeed, it seems that almost an inverse relationship exists, in that disease levels tend to be elevated where silica levels are lower (e.g., in areas where anthracite is mined). Recently, some understanding of this paradox has been gained through studies of particle characteristics. These studies indicate that not only the quantity of silica present in the dust (as measured conventionally using infrared spectrometry or x-ray diffraction), but also the bioavailability of the surface of the silica particles may be related to toxicity. For example, clay coating (occlusion) may play an important modifying role. Another important factor under current investigation concerns surface charge in the form of free radicals and the effects of “freshly fractured” versus “aged” silica-containing dusts.
Surveillance and epidemiology
The prevalence of CWP among underground miners varies with the kind of job, tenure and age. A recent study of US coal miners revealed that from 1970 to 1972 about 25 to 40% of working coal miners had category 1 or greater small rounded opacities after 30 or more years in mining. This prevalence reflects exposure to levels of 6 mg/m3 or more of respirable dust among coal face workers prior to that time. The introduction of a dust limit of 3 mg/m3 in 1969, with a reduction to 2 mg/m3 in 1972 has led to a decline in disease prevalence to about half of the former levels. Declines related to dust control have been noted elsewhere, for example, in the United Kingdom and Australia. Unfortunately, these gains have been counterbalanced by temporal increases in prevalence elsewhere.
An exposure-response relationship for prevalence or incidence of CWP and dust exposure has been demonstrated in a number of studies. These have shown that the primary significant dust exposure variable is exposure to mixed mine dust. Intensive studies by British researchers failed to disclose any major influence of silica exposure, as long as the percentage of silica was less than about 5%. Coal rank (percentage carbon) is another important predictor of CWP development. Studies in the United States, the United Kingdom, Germany and elsewhere have given clear indications that the prevalence and incidence of CWP increases markedly with coal rank, these being substantially greater where anthracite (high rank) coal is mined. No other environmental variables have been found to exert any major effects on CWP development. Miner age appears to have some bearing on disease development, since older miners appear to be at increased risk. However, it is not entirely clear whether this implies that older miners are more susceptible, whether it is a residence time effect, or is simply an artefact (the age effect might reflect underestimation of exposure estimates for older miners, for example). Cigarette smoking does not appear to increase the risk of CWP development.
Research in which miners were followed-up with chest radiographs every five years shows that the risk of developing PMF over the five years is clearly related to the category of CWP as revealed on the initial chest x ray. Since the risk at category 2 is much greater than that at category 1, conventional wisdom at one time was that miners should be prevented from reaching category 2 if at all possible. However, in most mines there are usually many more miners with category 1 CWP compared to category 2. Thus, the lower risk for category 1 compared to category 2 is offset somewhat by the larger numbers of miners with category 1. On this showing, it has become clear that all pneumoconiosis should be prevented.
Mortality
Miners as a group have been observed to have increased risk of death from non-malignant respiratory diseases, and there is evidence that the mortality among miners with CWP is somewhat increased over those of similar age without the disease. However, the effect is smaller than the excess seen for miners with PMF (see below).
Prevention
The only protection against CWP is minimization of dust exposure. If possible, this should be achieved by dust suppression methods, such as ventilation and water sprays, rather than by respirator use or administrative controls, for example, worker rotation. In this respect, there is now good evidence that regulatory actions in some countries to reduce the level of dust, taken around the 1970s, has resulted in greatly reduced levels of disease. Transfer of workers with early signs of CWP to less dusty jobs is a prudent action, although there is little practical evidence that such programmes have succeeded in preventing disease progression. For this reason, dust suppression must remain the primary method of disease prevention.
Ongoing, aggressive monitoring of dust exposure and the conscious exertion of control efforts can be supplemented by health screening surveillance of miners. If miners are found to develop dust-related diseases, efforts at exposure control should be intensified throughout the workplace and miners with dust effects should be offered work in low-dust areas of the mine environment.
Treatment
Although several forms of treatment have been tried, including aluminium powder inhalation, and the administration of tetrandine, no treatment is known that effectively reverses or slows the fibrotic process in the lung. Currently, primarily in China, but elsewhere also, whole-lung lavage is being tried with the intent of reducing the total lung dust burden. Although the procedure can result in the removal of a considerable amount of dust, its risks, benefits and role in the management of miners’ health are unclear.
In other respects, treatment should be directed towards preventing complications, maximizing the miners’ functional status and alleviating their symptoms, whether due to CWP or to other, concomitant respiratory diseases. In general, miners who develop dust-induced lung diseases should evaluate their current dust exposures and utilize the resources of government and labour organizations to find the avenues available to reduce all adverse respiratory exposures. For miners who smoke, smoking cessation is an initial step in personal exposure management. Prevention of infectious complications of chronic lung disease with available pneumococcal and yearly influenza vaccines is suggested. Early investigation of symptoms of lung infection, with particular attention to mycobacterial disease, is also recommended. The treatments for acute bronchitis, bronchospasm and congestive heart failure among miners are similar to those for patients without dust-related disease.
Progressive Massive Fibrosis
PMF, sometimes referred to as complicated pneumoconiosis, is diagnosed when one or more large fibrotic lesions (whose definition depends on the mode of detection) are present in one or both lungs. As its name implies, PMF often becomes more severe over time, even in the absence of additional dust exposure. It can also develop after dust exposure has ceased, and may often cause disability and premature mortality.
Pathology
PMF lesions may be unilateral or bilateral, and are most often found in the upper or middle lobes of the lung. The lesions are formed of collagen, reticulin, coal mine dust and dust-laden macrophages, while the centre may contain a black liquid which cavitates on occasion. US pathology standards require the lesions to be 2 cm in size or larger to be identified as PMF entities in surgical or autopsy specimens.
Radiology
Large opacities >>1 cm) on the radiograph, coupled with a history of extensive coal mine dust exposure, are taken to imply the presence of PMF. However, it is important that other diseases such as lung cancer, tuberculosis and granulomas be considered. Large opacities are usually seen on a background of small opacities, but development of PMF from a category 0 profusion has been noted over a five-year period.
Clinical aspects
Diagnostic possibilities for each individual miner with large chest opacities must be appropriately evaluated. Clinically stable miners with bilateral lesions in the typical upper-lung distribution and with pre-existing simple CWP may present little diagnostic challenge. However, miners with progressive symptoms, risk factors for other disorders (e.g., tuberculosis), or atypical clinical features should undergo a thorough appropriate examination before the diagnostician attributes the lesions to PMF.
Dyspnoea and other respiratory symptoms often accompany PMF, but may not necessarily be due to the disease itself. Congestive heart failure (due to pulmonary hypertension and cor pulmonale) is a not infrequent complication.
Disease mechanisms
Despite extensive research, the actual cause of PMF development remains unclear. Over the years, various hypotheses have been proposed, but none is fully satisfactory. One prominent theory was that tuberculosis played a role. Indeed, tuberculosis is often present in miners with PMF, particularly in the developing countries. However, PMF has been found to develop in miners in whom there was no sign of tuberculosis, and tuberculin reactivity has not been found to be elevated in miners with pneumoconiosis. Despite investigation, consistent evidence of the role of the immune system in PMF development is lacking.
Surveillance and epidemiology
As with CWP, PMF levels have been declining in countries which have strict dust control regulations and programmes. A recent study of US miners revealed that about 2% of coal miners working underground had PMF after 30 or more years in mining (although this figure may have been biased by affected miners leaving the work force).
Exposure-response investigations of PMF have shown that exposure to coal mine dust, category of CWP, coal rank and age are the primary determinants of disease development. As with CWP, epidemiological studies have found no major effect of silica dust. Although it was thought at one time that PMF developed only on a background of the small opacities of CWP, recently this has been found not to be the case. Miners with an initial chest x ray showing category 0 CWP have been shown to develop PMF over five years, with the risk increasing with their cumulative dust exposure. Also, miners may develop PMF after cessation of dust exposure.
Mortality
PMF leads to premature mortality, the prognosis worsening with increasing stage of the disease. A recent study showed that miners with category C PMF had only one-fourth the rate of survival over 22 years compared to miners with no pneumoconiosis. This effect was manifested over all age groups.
Prevention
Avoidance of dust exposure is the only way to prevent PMF. Since the risk of its development increases sharply with increasing category of simple CWP, a strategy for secondary prevention of PMF is for miners to undergo periodic chest x rays and to terminate or reduce their exposure if simple CWP is detected. Although this approach appears valid and has been adopted in certain jurisdictions, its effectiveness has not been evaluated systematically.
Treatment
There is no known treatment for PMF. Medical care should be organized around ameliorating the condition and associated lung illnesses, while protecting against infectious complications. Although maintaining functional stability may be more difficult in patients with PMF, in other respects, management is similar to simple CWP.
Obstructive Lung Disease
There is now consistent and convincing evidence of a relationship between lung function loss and dust exposure. Various studies in different countries have looked at the influence of dust exposure on absolute values of, and temporal changes in, measurements of ventilatory function, such as forced expiratory volume in one second (FEV1), forced vital capacity (FVC) and flow rates. All have found evidence that dust exposure leads to a reduction in lung function, and the results have been strikingly similar for several recent British and US investigations. These indicate that over the course of a year, dust exposure at the coal face brings about, on average, a reduction in lung function equivalent to smoking half a pack of cigarettes each day. The studies also demonstrate that effects vary, and a given miner may develop effects equal to, or worse than, those expected from cigarette smoking, particularly if the individual has experienced higher dust exposures.
The effects of dust exposure have been found in both those who have never smoked and in current smokers. Moreover, there is no evidence that smoking exacerbates the dust exposure effect. Rather, studies have generally shown a slightly smaller effect in current smokers, a result that may be due to healthy worker selection. It is important to note that the relationship between dust exposure and ventilatory decline appears to exist independently of pneumoconiosis. That is, it is not a requirement that pneumoconiosis be present for there to be reduced lung function. To the contrary, it appears rather that the inhaled dust can act along multiple pathways, leading to pneumoconiosis in some miners, to obstruction in others and to multiple outcomes in yet others. In contrast to miners with CWP alone, miners with respiratory symptoms have significantly lower lung function, after standardization for age, smoking, dust exposure and other factors.
Recent work on ventilatory function changes has involved the exploration of longitudinal changes. The results indicate that there may be a non-linear trend of decline over time in new miners, a high initial rate of loss being followed by a more moderate decline with continued exposure. Furthermore, there is evidence that miners who react to the dust may choose, if possible, to remove themselves from the heavier exposures.
Chronic Bronchitis
Respiratory symptoms, such as chronic cough and phlegm production, are a frequent consequence of work in coal mining, most studies showing an excess prevalence compared to non-exposed control groups. Moreover, the prevalence and incidence of respiratory symptoms has been shown to increase with cumulative dust exposure, after taking into account age and smoking. The presence of symptoms appears to be associated with a reduction in lung function over and above that due to dust exposure and other putative causes. This suggests that dust exposure may be instrumental in initiating certain disease processes that then progress regardless of further exposure. A relationship between bronchial gland size and dust exposure has been demonstrated pathologically, and it has been found that mortality from bronchitis and emphysema increases with increasing cumulative dust exposure.
Emphysema
Pathological studies have repeatedly found an excess of emphysema in coal miners compared to control groups. Moreover, the degree of emphysema has been found to be related both to the amount of dust in the lungs and to pathological assessments of pneumoconiosis. Furthermore, it is important to recognize that there is evidence that the presence of emphysema is related to dust exposure and to the percentage of predicted FEV1. Hence, these results are consistent with the view that dust exposure can lead to disability through causing emphysema.
The form of emphysema most clearly associated with coal mining is focal emphysema. This consists of zones of enlarged air spaces, 1 to 2 mm in size, adjacent to dust macules surrounding the respiratory bronchioles. The current thinking is that the emphysema is formed from tissue destruction, rather than from distension or dilation. Apart from focal emphysema, there is evidence that centriacinar emphysema has an occupational origin, and that total emphysema, (i.e., the extent of all types) is correlated with tenure in mining, in those who have never smoked as well as in smokers. There is no evidence that smoking potentiates the dust exposure/emphysema relationship. However, there are indications of an inverse relationship between the silica content of lungs and the presence of emphysema.
The issue of emphysema has long been controversial, with some stating that selection bias and smoking make interpretation of pathological studies difficult. In addition, some consider that focal emphysema has only trivial effects on lung function. However, pathological studies undertaken since the 1980s have been responsive to earlier criticisms, and indicate that the effect of dust exposure may be more significant for miners’ health than previously thought. This point of view is supported by recent findings that mortality from bronchitis and emphysema is related to cumulative dust exposure.
Silicosis
Silicosis, though associated more with industries other than coal mining, can occur in coal miners. In underground mines, it is found most frequently in workers in certain jobs where exposure to pure silica typically occurs. Such workers include roof bolters, who drill into the ceiling rock, which can often be sandstone or other rock with high silica content; motormen, drivers of rail transport who are exposed to the dust generated by sand placed on the tracks to lend traction; and rock drillers, who are involved in mine development. Rock drillers at surface coal mines have been shown to be at particular risk in the United States, with some developing acute silicosis after only a few years of exposure. Based on pathological evidence, as noted below, some degree of silicosis may afflict many more coal miners than just those working the jobs noted above.
Silicotic nodules in coal miners are similar in nature to those observed elsewhere, and consist of a whorled pattern of collagen and reticulin. One large autopsy study has revealed that about 13% of coal miners had silicotic nodules in their lungs. Although one job, (that of motorman) was notable for having a much higher prevalence of silicotic nodules (25%), there was little variation in the prevalence among miners in other jobs, suggesting that the silica in the mixed mine dust was responsible.
Silicosis cannot be reliably differentiated from coal workers’ pneumoconiosis on a radiograph. However, there is some evidence that the larger type of small opacities (type r) are indicative of silicosis.
Rheumatoid Pneumoconiosis
Rheumatoid pneumoconiosis, one variant of which is called Caplan’s syndrome, is the term used for a condition affecting dust-exposed workers who develop multiple large radiographic shadows. Pathologically, these lesions resemble rheumatoid nodules rather than PMF lesions, and often arise over a short time interval. Active arthritis or the presence of circulating rheumatoid factor are generally found, but occasionally are absent.
Lung Cancer
Included in the occupational exposures suffered by coal miners are a number of substances that are potential carcinogens. Some of these are silica and benzo(a)pyrenes. Yet, there is no clear evidence of an excess of deaths from lung cancer in coal miners. One obvious explanation for this is that coal miners are forbidden to smoke underground because of the danger of fires and explosions. However, the fact that no exposure-response relationship between lung cancer and dust exposure has been detected suggests that coal mine dust is not a major cause of lung cancer in the industry.
Regulatory Limits on Dust Exposure
The World Health Organization (WHO) has recommended a “tentative health-based exposure limit” for respirable coal mine dust (with less than 6% respirable quartz) ranging from 0.5 to 4 mg/m3. WHO suggests a 2 in 1,000 risk of PMF over a working lifetime as a criterion, and recommends that mine-based environmental factors, including coal rank, percentage of quartz and particle size should be taken into account when setting limits.
Currently, among the major coal-producing countries, limits are based on regulating coal dust alone (e.g., 3.8 mg/m3 in the United Kingdom, 5 mg/m3 in Australia and Canada) or on regulating a mixture of coal and silica as in the United States (2 mg/m3 when the per cent quartz is 5 or less, or (10 mg/m3)/per cent SiO2), or in Germany (4 mg/m3 when the per cent quartz is 5 or less, or 0.15 mg/m3 otherwise), or on regulating pure quartz (e.g., Poland, with a 0.05 mg/m3 limit).
Asbestos-Related Diseases
Historical Perspective
Asbestos is a term used to describe a group of naturally occurring fibrous minerals which are very widely distributed in rock outcrops and deposits throughout the world. Exploitation of the tensile and heat-resistant properties of asbestos for human use dates from ancient times. For instance, in the third century BC asbestos was used to strengthen clay pots in Finland. In classic times, shrouds woven from asbestos were used to preserve the ashes of the famous dead. Marco Polo returned from his travels in China with descriptions of a magic material which could be manufactured into a flame resistant cloth. By the early years of the nineteenth century, deposits were known to exist in several parts of the world, including the Ural Mountains, northern Italy and other Mediterranean areas, in South Africa and in Canada, but commercial exploitation only started in the latter half of the nineteenth century. By this time, the industrial revolution created not only the demand (such as that of insulating the steam engine) but also facilitated production, with mechanization replacing hand cobbing of fibre from the parent rock. The modern industry began in Italy and the United Kingdom after 1860 and was boosted by the development and exploitation of the extensive deposits of chrysotile (white) asbestos in Quebec (Canada) in the 1880s. Exploitation of the also extensive deposits of chrysotile in the Ural mountains was modest until the 1920s. The long thin fibres of chrysotile were particularly suitable for spinning into cloth and felts, one of the early commercial uses for the mineral. The exploitation of the deposits of crocidolite (blue) asbestos of the northwest Cape, South Africa, a fibre more water-resistant than chrysotile and better suited to marine use, and of the amosite (brown) asbestos deposits, also found in South Africa, started in the early years of this century. Exploitation of the Finnish deposits of anthophyllite asbestos, the only important commercial source of this fibre, took place between 1918 and 1966, while the deposits of crocidolite in Wittenoom, Western Australia, were mined from 1937 to 1966.
Fibre Types
The asbestos minerals fall into two groups, the serpentine group which includes chrysotile, and the amphiboles, which include crocidolite, tremolite, amosite and anthophyllite (figure 1). Most ore deposits are heterogeneous mineralogically, as are most of the commercial forms of the mineral (Skinner, Roos and Frondel 1988). Chrysotile and the various amphibole asbestos minerals differ in crystalline structure, in chemical and surface characteristics and in the physical characteristics of their fibres, usually described in terms of the length-to-diameter (or aspect) ratio. They also differ in characteristics which distinguish commercial use and grade. Pertinent to the current discussion is the evidence that the different fibres differ in their biological potency (as considered below in the sections on various diseases).
Figure 1. Asbestos fibre types.

Seen on election microscopy together with energy dispersive x-ray spectra which enables identification of individual fibres. Courtesy of A. Dufresne and M. Harrigan, McGill University.
Commercial Production
The growth of commercial production, illustrated in figure 2, was slow in the early years of this century. For instance, Canadian production exceeded 100,000 short tons per annum for the first time in 1911 and 200,000 tons in 1923. Growth between the two World Wars was steady, increased considerably to meet the demands of the Second World War and spectacularly to meet peacetime demands (including those of the cold war) to reach a peak in 1976 of 5,708,000 short tons (Selikoff and Lee 1978). After this, production faltered as the ill-health effects of exposure became a matter of increasing public concern in North America and Europe and remain at approximately 4,000,000 short tons per annum up to 1986, but decreased further in the 1990s. There was also a shift in the uses and sources of fibre in the 1980s; in Europe and North America demand declined as substitutes for many applications were introduced, while on the African, Asian and South American continents, demand for asbestos increased to meet the needs of a cheap durable material for use in construction and in water reticulation. By 1981, Russia had become the world’s major producer, with an increase in the commercial exploitation of large deposits in China and Brazil. In 1980, it was estimated that a total of over 100 million tons of asbestos had been mined worldwide, 90% of which was chrysotile, approximately 75% of which came from 4 chrysotile mining areas, located in Quebec (Canada), Southern Africa and the central and southern Ural Mountains. Two to three per cent of the world’s total production was crocidolite, from the Northern Cape, South Africa, and from Western Australia, and another 2 to 3% was amosite, from the Eastern Transvaal, South Africa (Skinner, Ross and Frondel 1988).
Figure 2. World production of asbestos in thousands of tons 1900-92
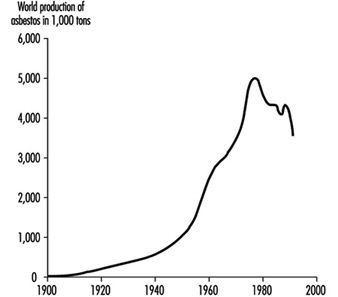
Asbestos-Related Diseases and Conditions
Like silica, asbestos has the capability of evoking scarring reactions in all biological tissue, human and animal. In addition, asbestos evokes malignant reactions, adding a further element to the concern for human health, as well as a challenge to science as to how asbestos exerts its ill effects. The first asbestos-related disease to be recognized, diffuse interstitial pulmonary fibrosis or scarring, later called asbestosis, was the subject of case reports in the United Kingdom in the early 1900s. Later, in the 1930s, case reports of lung cancer in association with asbestosis appeared in the medical literature though it was only over the next several decades that the scientific evidence was gathered establishing that asbestos was the carcinogenic factor. In 1960, the association between asbestos exposure and another much less common cancer, malignant mesothelioma, which involves the pleura (a membrane that covers the lung and lines the chest wall) was dramatically brought to attention by the report of a cluster of these tumours in 33 individuals, all of whom worked or lived in the asbestos mining area of the Northwest Cape (Wagner 1996). Asbestosis was the target of the dust control levels introduced and implemented with increasing rigour in the 1960s and 1970s, and in many industrialized countries, as the frequency of this disease decreased, asbestos-related pleural disease emerged as the most frequent manifestation of exposure and the condition which most frequently brought exposed subjects to medical attention. Table 1 lists diseases and conditions currently recognized as asbestos-related. The diseases in bold type are those most frequently encountered and for which a direct causal relationship is well established, while for the sake of completeness, certain other conditions, for which the relationship is less well established, are also listed (see footnote to Table 16) and the sections which follow in the text below that expand upon the various disease headings).
Table 1. Asbestos-related diseases and conditions
| Pathology | Organ(s) affected | Disease/condition1 |
| Non-malignant | Lungs Pleura Skin | Asbestosis (diffuse interstitial fibrosis) Small airway disease2 (fibrosis limited to the peri-bronchiolar region) Chronic airways disease3 Pleural plaques Viscero-parietal reactions, including benign pleural effusion, diffuse pleural fibrosis and rounded atelectasis Asbestos corns4 |
| Malignant | Lungs Pleura Other mesothelium-lined cavities Gastrointestinal tract5 Other5 | Lung cancer (all cell types) Cancer of larynx Mesothelioma of pleura Mesothelioma of the peritoneum, pericardium and scrotum (in decreasing frequency of occurrence) Cancer of stomach, oesophagus, colon, rectum Ovary, gall bladder, bile ducts, pancreas, kidney |
1 The diseases or conditions indicated in bold type are those most frequently encountered and the ones for which a causal relationship is well established and/or generally recognized.
2 Fibrosis in the walls of the small airways of the lung (including the membranous and respiratory bronchioles) is thought to represent the early lung parenchymal response to retained asbestos (Wright et al. 1992) which will progress to asbestosis if exposure continues and/or is heavy, but if exposure is limited or light, the lung response may be limited to these areas (Becklake in Liddell & Miller 1991).
3 Included are bronchitis, chronic obstructive pulmonary disease (COPD) and emphysema. All have been shown to be associated with work in dusty environments. The evidence for causality is reviewed in the section Chronic Airways Diseases and Becklake (1992).
4 Related to direct handling of asbestos and of historical rather than current interest.
5 Data not consistent from all studies (Doll and Peto 1987); some of the highest risks were reported in a cohort of over 17,000 American and Canadian asbestos insulation workers (Selikoff 1990), followed from January 1, 1967 to December 31, 1986 in whom exposure had been particularly heavy.
Sources: Becklake 1994; Liddell and Miller 1992; Selikoff 1990; Doll and Peto in Antman and Aisner 1987; Wright et al. 1992.
Uses
Table 2 lists the major sources, products and uses of the asbestos minerals.
Table 2. Main commercial sources, products and uses of asbestos
| Fibre type | Location of major deposits | Commercial products and/or uses |
| Chrysotile (white) |
Russia, Canada (Québec, also British Columbia, Newfoundland), China (Szechwan province); Mediterranean countries (Italy, Greece, Corsica, Cyprus); Southern Africa (South Africa, Zimbabwe, Swaziland); Brazil; smaller deposits in United States (Vermont, Arizona, California) and in Japan | Construction materials (tiles, shingles, gutters and cisterns; roofing, sheeting, and siding) Pressure and other pipes Fire proofing (marine and other) Insulation and sound proofing Reinforced plastic products (fan blades, switch gear) Friction materials usually in combination with resins in brakes, clutches, other Textiles (used in belts, clothing, casing, fire barriers, autoclaves, yarns and packing) Paper products (used in millboard, insulators, gaskets, roof felt, wall coverings, etc.) Floats in paints, coatings and welding rods |
| Crocidolite (blue) |
South Africa (Northwest Cape, Eastern Transvaal), Western Australia1 | Used mainly in combination in cement products (in particular pressure pipes) but also in many of the other products listed above |
| Amosite (brown) |
South Africa (Northern Transvaal)1 | Used mainly in cement, thermal insulation and roofing products particularly in the United States2 , but also in combination in many of the products listed under chrysotile |
| Anthophyllite | Finland1 | Filler in the rubber, plastics and chemical industries |
| Tremolite | Italy, Korea and some Pacific Islands; mined on a small scale in Turkey, China and elsewhere; contaminates the ore bearing rock in some asbestos, iron, talc and vermiculite mines; also found in agricultural soils in the Balkan Peninsula and in Turkey | Used as a filler in talc; may or may not be removed in processing the ore so it may appear in end products |
| Actinolite | Contaminates amosite, and less often, chrysotile, talc and vermiculite deposits | Not usually exploited commercially |
1 A list such as this is obviously not comprehensive and the readers should consult the sources cited and other chapters in this Encyclopedia for more complete information.
2 No longer in operation.
Sources: Asbestos Institute (1995); Browne (1994); Liddell and Miller (1991); Selikoff and Lee (1978); Skinner et al (1988).
Though necessarily incomplete, this table emphasizes that:
- Deposits are found in many parts of the world, most of which have been exploited non-commercially or commercially in the past, and some of which are currently commercially exploited.
- There are many manufactured products in current or past use which contain asbestos, particularly in the construction and transport industries.
- Disintegration of these products or their removal carries with it the risk of the resuspension of fibres and of renewed human exposure.
A figure of over 3,000 has been commonly quoted for the number of uses of asbestos and no doubt led to asbestos being dubbed the “magic mineral” in the 1960s. A 1953 industry list contains as many as 50 uses for raw asbestos, in addition to its use in the manufacture of the products listed in Table 17, each of which has many other industrial applications. In 1972, the consumption of asbestos in an industrialized country like the United States was attributed to the following categories of product: construction (42%); friction materials, felts, packings and gaskets (20%); floor tiles (11%); paper (9%); insulation and textiles (3%) and other uses (15%) (Selikoff and Lee 1978). By contrast, a 1995 industry list of the main product categories shows major redistribution on a worldwide basis as follows: asbestos cement (84%); friction materials (10%); textiles (3%); seals and gaskets (2%); and other uses (1%) (Asbestos Institute 1995).
Occupational Exposures, Past and Current
Occupational exposure, certainly in industrialized countries, has always been and is still the most likely source of human exposure (see Table 17 and the references cited in its footnote; other sections of this Encyclopaedia contain further information). There have, however, been major changes in industrial processes and procedures aimed at diminishing the release of dust into the working environment (Browne 1994; Selikoff and Lee 1978). In countries with mining operations, milling usually takes place at the minehead. Most chrysotile mines are open cast, while amphibole mines usually involve underground methods which generate more dust. Milling involves separating fibre from rock by means of mechanized crushing and screening, which were dusty processes until the introduction of wet methods and/or enclosure in most mills during the 1950s and 1960s. The handling of waste was also a source of human exposure, as was transporting bagged asbestos, whether it involved loading and unloading trucks and railcars or work on the dockside. These exposures have diminished since the introduction of leak-proof bags and the use of sealed containers.
Workers have had to use raw asbestos directly in packing and lagging, particularly in locomotives, and in spraying walls, ceilings and airducts, and in the marine industry, deckheads and bulkheads. Some of these uses have been phased out voluntarily or have been banned. In the manufacture of asbestos cement products, exposure occurs in receiving and opening bags containing raw asbestos, in preparing the fibre for mixing in the slurry, in machining end-products and in dealing with waste. In the manufacture of vinyl tiles and flooring, asbestos was used as a reinforcing and filler agent to blend with organic resins, but has now largely been replaced by organic fibre in Europe and North America. In the manufacture of yarns and textiles, exposure to fibre occurs in receiving, preparing, blending, carding, spinning, weaving and calendaring the fibre—processes which were until recently dry and potentially very dusty. Dust exposure has been considerably reduced in modern plants through use of a colloidal suspension of fibre extruded through a coagulant to form wet strands for the last-mentioned three processes. In the manufacture of asbestos paper products, human exposure to asbestos dust is also most likely to occur in the reception and preparation of the stock mix and in cutting the final products which in the 1970s contained from 30 to 90% asbestos. In the manufacture of asbestos friction products (dry mix-moulded, roll-formed, woven or endless wound) human exposure to asbestos dust is also most likely to occur during the initial handling and blending processes as well as in finishing the end product, which in the 1970s contained from 30 to 80% of asbestos. In the construction industry, prior to regular use of appropriate exhaust ventilation (which came in the 1960s), the high-speed power sawing, drilling and sanding of asbestos-containing boards or tiles led to the release of fibre-containing dust close to the operator’s breathing zone, particularly when such operations were conducted in closed spaces (for instance in high-rise buildings under construction). In the period after the Second World War, a major source of human exposure was in the use, removal or replacement of asbestos-containing materials in the demolition or refurbishing of buildings or ships. One of the chief reasons for this state of affairs was the lack of awareness, both of the composition of these materials (i.e., that they contained asbestos) and that exposure to asbestos could be harmful to health. Improved worker education, better work practices and personal protection have reduced the risk in the 1990s in some countries. In the transport industry, sources of exposure were the removal and replacement of lagging in locomotive engines and of braking material in trucks and cars in the automobile repair industry. Other sources of past exposure leading to, in particular, pleural disease, continue to attract notice, even in the 1990s, usually on the basis of case reports, for instance those describing workers using asbestos string in the manufacture of welding rods, in the formation of asbestos rope for grouting furnaces and maintaining underground mine haulage systems.
Other Sources of Exposure
Exposure of individuals engaged in trades which do not directly involve use or handling of asbestos but who work in the same area as those who do deal with it directly is called para-occupational (bystander) exposure. This has been an important source of exposure not only in the past but also for cases presenting for diagnosis in the 1990s. Workers involved include electricians, welders and carpenters in the construction and in the ship building or repair industries; maintenance personnel in asbestos factories; fitters, stokers and others in power stations and ships and boiler houses where asbestos lagging or other insulation is in place, and maintenance personnel in post-war high-rise buildings incorporating various asbestos-containing materials. In the past, domestic exposure occurred primarily from dust-laden workclothes being shaken or laundered at home, the dust so released becoming entrapped in carpets or furnishings and resuspended into the air with the activities of daily living. Not only could levels of airborne fibre reach levels as high as 10 fibre per millilitre (f/ml), that is, ten times the occupational exposure limit proposed by a WHO consultation (1989) of 1.0 f/ml but the fibres tended to remain airborne for several days. Since the 1970s, the practice of retaining all work clothes at the worksite for laundering has been widely but not universally adopted. In the past also, residential exposure occurred from contamination of air from industrial sources. For instance, increased levels of airborne asbestos have been documented in the neighbourhood of mines and asbestos plants and are determined by production levels, emission controls and weather. Given the long lag time for, in particular, asbestos-related pleural disease, such exposures are still likely to be responsible for some cases presenting for diagnosis in the 1990s. In the 1970s and 1980s, with the increase in public awareness of both the ill-health consequences of asbestos exposure and of the fact that asbestos containing materials are used extensively in modern construction (particularly in the friable form used for spray-on applications to walls, ceilings and ventilation ducts), a major cause of concern centred on whether, as such buildings age and are subject to daily wear and tear, asbestos fibres may be released into the air in sufficient numbers to become a threat to the health of those working in modern high-rise buildings (see below for risk estimates). Other sources of contamination of the air in urban areas include the release of fibre from brakes of vehicles and rescattering of fibres released by passing vehicles (Bignon, Peto and Saracci 1989).
Non-industrial sources of environmental exposure include naturally occurring fibres in soils, for instance in eastern Europe, and in rock outcrops in the Mediterranean region, including Corsica, Cyprus, Greece and Turkey (Bignon, Peto and Saracci 1989). An additional source of human exposure results from the use of tremolite for whitewash and stucco in Greece and Turkey, and according to more recent reports, in New Caledonia in the South Pacific (Luce et al. 1994). Furthermore, in several rural villages in Turkey, a zeolite fibre, erionite, has been found to be used both in stucco and in domestic construction and has been implicated in mesothelioma production (Bignon, Peto and Saracci 1991). Finally, human exposure may occur through drinking water, mainly from natural contamination, and given the widespread natural distribution of the fibre in outcrops, most water sources contain some fibre, levels being highest in mining areas (Skinner, Roos and Frondel 1988).
Aetiopathology of Asbestos-Related Disease
Fate of inhaled fibres
Inhaled fibres align themselves with the airstream and their capability of penetrating into the deeper lung spaces depends on their dimension, fibres of 5mm or less in aerodynamic diameter showing an over 80% penetration, but also less than 10 to 20% retention. Larger particles may impact in the nose and in major airways at bifurcations, where they tend to collect. Particles deposited in the major airways are cleared by the action of ciliated cells and are transported up the mucus escalator. Individual differences associated with what appears to be the same exposure are due, at least in part, to differences between individuals in the penetration and retention of inhaled fibres (Bégin, Cantin and Massé 1989). Small particles deposited beyond the major airways are phagocytosed by alveolar macrophages, scavenger cells which ingest foreign material. Longer fibres, that is, those over 10mm, often come under attack by more than one macrophage, are more likely to become coated and to form the nucleus of an asbestos body, a characteristic structure recognized since the early 1900s as a marker of exposure (see figure 3). Coating a fibre is considered to be part of the lungs’ defense to render it inert and non-immunogenic. Asbestos bodies are more likely to form on amphibole than on chrysotile fibres, and their density in biological material (sputum, bronchoalveolar lavage, lung tissue) is an indirect marker of lung burden. Coated fibres may persist in the lung for long periods, to be recovered from sputum or bronchoalveolar lavage fluid up to 30 years after last exposure. Clearance of non-coated fibres deposited in the lung’s parenchyma is towards the lung periphery and subpleural regions, and then to lymph nodes at the root of the lung.
Figure 3. Asbestos body
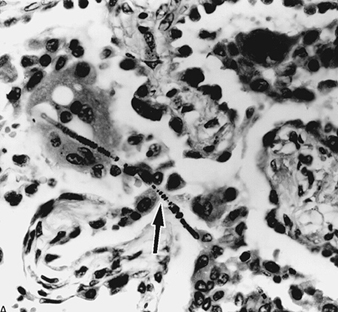
Magnification x 400, seen on microscopic section of the lung as a slightly curved elongated structure with a finely beaded iron protein coat. The asbestos fibre itself can be identified as the thin line near one end of the asbestos body (arrow). Source: Fraser et al. 1990
Theories to explain how fibres evoke the various pleural reactions associated with asbestos exposure include:
- direct penetration into the pleural space and drainage with the pleural fluid to pores in the pleura lining the chest wall
- release of mediators into the pleural space from subpleural lymphatic collections
- retrograde flow from lymph nodes at the root of the lung to the parietal pleura (Browne 1994)
There may also be retrograde flow via the thoracic duct to the abdominal lymph nodes to explain the occurrence of peritoneal mesothelioma.
Cellular effects of inhaled fibres
Animal studies indicate that the initial events which follow asbestos retention in the lung include:
- an inflammatory reaction, with accumulation of white blood cells followed by a macrophagic alveolitis with release of fibronectin, growth factor and various neutrophil chemotactic factors and, over time, the release of superoxide ion and
- proliferation of alveolar, epithelial, interstitial and endothelial cells (Bignon, Peto and Saracci 1989).
These events are reflected in the material recovered by bronchoalveolar lavage in animals and humans (Bégin, Cantin and Massé 1989). Both fibre dimensions and their chemical characteristics appear to determine biological potency for fibrogenesis, and these characteristics, in addition to surface properties, are also thought to be important for carcinogenesis. Long, thin fibres are more active than short ones, although the activity of the latter cannot be discounted, and amphiboles are more active than chrysotile, a property attributed to their greater biopersistence (Bégin, Cantin and Massé 1989). Asbestos fibres may also affect the human immune system and change the circulating population of blood lymphocytes. For instance, human cell mediated immunity to cell antigens (such as is exhibited in a tuberculin skin test) may be impaired (Browne 1994). In addition, since asbestos fibres appear to be capable of inducing chromosome abnormality, the view has been expressed that they can also be considered capable of inducing as well as promoting cancer (Jaurand in Bignon, Peto and Saracci 1989).
Dose versus exposure response relationships
In biological sciences such as pharmacology or toxicology in which dose-response relationships are used to estimate the probability of desired effects or the risk of undesired effects, a dose is conceptualized as the amount of agent delivered to and remaining in contact with the target organ for sufficient time to evoke a reaction. In occupational medicine, surrogates for dose, such as various measures of exposure, are usually the basis for risk estimates. However, exposure-response relationships can usually be demonstrated in workforce-based studies; the most appropriate exposure measure may, however, differ between diseases. Somewhat disconcerting is the fact that although exposure-response relationships will differ between workforces, these differences can be explained only in part by the fibre, particle size and industrial process. Nevertheless, such exposure-response relationships have formed the scientific basis for risk assessment and for setting permissible exposure levels, which were originally focused on controlling asbestosis (Selikoff and Lee 1978). As the prevalence and/or incidence of this condition has decreased, concern has switched to assure protection of human health against asbestos-related cancers. Over the last decade, techniques have been developed for the quantitative measurement of lung dust burden or biological dose directly in terms of fibres per gram of dry lung tissue. In addition, energy dispensive x-ray analysis (EDXA) permits precise characterization of each fibre by fibre type (Churg 1991). Though standardization of results between laboratories has not yet been achieved, comparisons of results obtained within a given laboratory are useful, and lung burden measurements have added a new tool for case evaluation. In addition, the application of these techniques in epidemiological studies has
- confirmed the biopersistence of amphibole fibres in the lung compared to chrysotile fibres
- identified fibre burden in the lungs of some individuals in whom exposure was forgotten, remote or thought to be unimportant
- demonstrated a gradient in lung burden associated with rural and urban residence and with occupational exposure and
- confirmed a fibre gradient in the lung dust burden associated with the major asbestos-related diseases (Becklake and Case 1994).
Asbestosis
Definition and history
Asbestosis is the name given to the pneumoconiosis consequent on exposure to asbestos dust. The term pneumoconiosis is used here as defined in the article “Pneumoconioses: Definitions”, of this Encyclopaedia as a condition in which there is “accumulation of dust in the lungs and tissue responses to the dust”. In the case of asbestosis, the tissue reaction is collagenous, and results in permanent alteration of the alveolar architecture with scarring. As early as 1898, the Annual Report of Her Majesty’s Chief Inspector of Factories contained reference to a lady factory inspector’s report on the adverse health consequences of asbestos exposure, and the 1899 Report contained details of one such case in a man who had worked for 12 years in one of the recently established textile factories in London, England. Autopsy revealed diffuse severe fibrosis of the lung and what subsequently came to be known as asbestos bodies were seen on subsequent histologic re-examination of the slides. Since fibrosis of the lung is an uncommon condition, the association was thought to be causal and the case was presented in evidence to a committee on compensation for industrial disease in 1907 (Browne 1994). Despite the appearance of reports of a similar nature filed by inspectors from the United Kingdom, Europe and Canada over the next decade, the role of exposure to asbestos in the genesis of the condition was not generally recognized until a case report was published in the British Medical Journal in 1927. In this report, the term pulmonary asbestosis was first used to describe this particular pneumoconiosis, and comment was made on the prominence of the associated pleural reactions, in contrast, for instance, to silicosis, the main pneumoconiosis recognized at the time (Selikoff and Lee 1978). In the 1930s, two major workforce-based studies carried out among textile workers, one in the United Kingdom and one in the United States, provided evidence of an exposure-response (and therefore likely causal) relationship between level and duration of exposure and radiographic changes indicative of asbestosis. These reports formed the basis of the first control regulations in the United Kingdom, promulgated in 1930, and the first threshold limit values for asbestos published by the American Conference of Government and Industrial Hygienists in 1938 (Selikoff and Lee 1978).
Pathology
The fibrotic changes which characterize asbestosis are the consequence of an inflammatory process set up by fibres retained in the lung. The fibrosis of asbestosis is interstitial, diffuse, tends to involve the lower lobes and peripheral zones preferentially and, in the advanced case, is associated with obliteration of the normal lung architecture. Fibrosis of the adjacent pleura is common. Nothing in the histological features of asbestosis distinguish it from interstitial fibrosis due to other causes, except the presence of asbestos in the lung either in the form of asbestos bodies, visible to light microscopy, or as uncoated fibres, most of which are too fine to be seen except by means of electron microscopy. Thus, the absence of asbestos bodies in images derived from light microscopy does not rule out either exposure or the diagnosis of asbestosis. At the other end of the spectrum of disease severity, the fibrosis may be limited to relatively few zones and affect mainly the peribronchiolar regions (see figure 4), giving rise to what has been called asbestos-related small airways disease. Again, except perhaps for more extensive involvement of membranous small airways, nothing in the histologic changes of this condition distinguishes it from small airways disease due to other causes (such as cigarette smoking or exposure to other mineral dusts) other than the presence of asbestos in the lung. Small airways disease may be the only manifestation of asbestos-related lung fibrosis or it may coexist with varying degrees of interstitial fibrosis, that is, asbestosis (Wright et al. 1992). Carefully considered criteria have been published for the pathological grading of asbestosis (Craighead et al. 1982). In general, the extent and intensity of the lung fibrosis relates to the measured lung dust burden (Liddell and Miller 1991).
Figure 4. Asbestos-related small airways disease
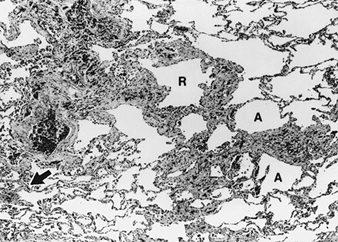
Peribronchiolar fibrosis and infiltration by inflammatory cells is seen on a histologic section of a respiratory bronchiole (R) and its distal divisions or alveolar ducts (A). The surrounding lung is mostly normal but with focal thickening of the interstitial tissue (arrow), representing early asbestosis. Source: Fraser et al. 1990
Clinical features
Shortness of breath, the earliest, most consistently reported and most distressing complaint, has led to asbestosis being called a monosymptomatic disease (Selikoff and Lee 1978). Shortness of breath precedes other symptoms which include a dry, often distressing cough, and chest tightness—which is thought to be associated pleural reactions. Late inspiratory rales or crackles which persist after coughing are heard, first in the axilla and over the lung bases, before becoming more generalized as the condition advances, and are thought to be due to the explosive opening of airways which close on expiration. Coarse rales and rhonchi, if present, are thought to reflect bronchitis either in response to working in a dusty environment, or due to smoking.
Chest imaging
Traditionally, the chest radiograph has been the most important single diagnostic tool for establishing the presence of asbestosis. This has been facilitated by the use of the ILO (1980) radiological classification, which grades the small irregular opacities that are characteristic of asbestosis on a continuum from no disease to the most advanced disease, both for severity (described as profusion on a 12-point scale from –/0 to 3/+) and extent (described as the number of zones affected). Despite between reader differences, even among those who have completed training courses in reading, this classification has proved particularly useful in epidemiological studies, and has also been used clinically. However, pathological changes of asbestosis can be present on lung biopsy in up to 20% of subjects with a normal chest radiograph. In addition, small irregular opacities of low profusion (e.g., 1/0 on the ILO scale) are not specific for asbestosis but can be seen in relation to other exposures, for instance to cigarette smoking (Browne 1994). Computer tomography (CT) has revolutionized the imaging of interstitial lung disease, including asbestosis, with high resolution computer tomography (HRCT) adding increased sensitivity to the detection of interstitial and pleural disease (Fraser et al. 1990). Characteristics of asbestosis which can be identified by HRCT include thickened interlobular (septal) and intralobular core lines, parenchymal bands, curvilinear subpleural lines and subpleural dependent densities, the first two being the most distinctive for asbestosis (Fraser et al. 1990). HRCT can also identify these changes in cases with pulmonary function deficit in whom the chest radiograph is inconclusive. Based on postmortem HRCT, thickened intralobular lines have been shown to correlate with peribronchiolar fibrosis, and thickened interlobular lines with interstitial fibrosis (Fraser et al. 1990). As yet, no standardized reading method has been developed for the use of HRCT in asbestos-related disease. In addition to its cost, the fact that a CT device is a hospital installation makes it unlikely that it will replace the chest radiograph for surveillance and epidemiological studies; its role will likely remain limited to individual case investigation or to planned studies intended to address specific issues. Figure 21 illustrates the use of chest imaging in the diagnosis of asbestos-related lung disease; the case shown exhibits asbestosis, asbestos-related pleural disease and lung cancer. Large opacities, a complication of other pneumoconioses, in particular silicosis, are unusual in asbestosis and are usually due to other conditions such as lung cancer (see the case described in figure 5) or rounded atelectasis.
Figure 5. Chest imaging in asbestos-related lung disease.
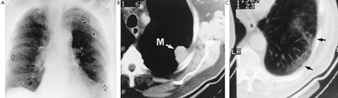
A posteroanterior chest radiograph (A) shows asbestosis involving both lungs and assessed as ILO category 1/1, associated with bilateral pleural thickening (open arrows) and a vaguely defined opacity (arrow heads) in the left upper lobe. On HRCT scan (B), this was shown to be a dense mass (M) abutting onto the pleura and transthoracic needle biopsy revealed an adenocarcinoma of the lung. Also on CT scan (C), at high attenuation pleural plaques can be seen (arrow heads) as well as a thin curvilinear opacity in the parenchyma underlying the plaques with interstitial abnormality in the lung between the opacity and the pleura. Source: Fraser et al. 1990
Lung function tests
Established interstitial lung fibrosis due to asbestos exposure, like established lung fibrosis due to other causes, is usually but not invariably associated with a restrictive lung function profile (Becklake 1994). Its features include reduced lung volumes, in particular vital capacity (VC) with preservation of the ratio of forced expiratory volume in 1second to forced vital capacity (FEV1/FVC%), reduced lung compliance, and impaired gas exchange. Air flow limitation with reduced FEV1/FVC may, however, also be present as a response to a dusty work environment or to cigarette smoke. In the earlier stages of asbestosis, when the pathological changes are limited to peribronchiolar fibrosis and even before small irregular opacities are evident on the chest radiograph, impairment of tests reflecting small airway dysfunction such as the Maximum Mid-expiratory Flow Rate may be the only sign of respiratory dysfunction. Responses to the stress of exercise may also be impaired early in the disease, with increased ventilation in relation to the oxygen requirement of the exercise (due to an increased breathing frequency and shallow breathing) and impaired O2 exchange. As the disease progresses, less and less exercise is required to compromise O2 exchange. Given that the asbestos-exposed worker may exhibit features of both a restrictive and an obstructive lung function profile, the wise physician interprets the lung function profile in the asbestos worker for what it is, as a measure of impairment, rather than as an aid to diagnosis. Lung functions, in particular vital capacity, provide a useful tool for the follow-up of subjects individually, or in epidemiological studies, for instance after exposure has ceased, to monitor the natural history of asbestosis or asbestos-related pleural disease.
Other laboratory tests
Bronchoalveolar lavage is increasingly used as a clinical tool in the investigation of asbestos-related lung disease:
- to rule out other diagnoses
- to assess the activity of the pulmonary reactions under study such as fibrosis or
- to identify the agent in the form of asbestos bodies or fibres.
It is also used to study disease mechanisms in humans and animals (Bégin, Cantin and Massé 1989). The uptake of Gallium-67 is used as a measure of the activity of the pulmonary process, and serum antinuclear antibodies (ANA) and rheumatoid factors (RF), both of which reflect the immunological status of the individual, have also been investigated as factors influencing disease progression, and/or accounting for between individual differences in response to what appears to be the same level and dose of exposure.
Epidemiology including natural history
The prevalence of radiological asbestosis documented in workforce-based surveys varies considerably and, as might be expected, these differences relate to differences in exposure duration and intensity rather than differences between workplaces. However, even when these are taken into account by restricting comparison of exposure response relationships to those studies in which exposure estimates were individualized for each cohort member and based on job history and industrial hygiene measurements, marked fibre and process related gradients are evident (Liddell and Miller 1991). For instance, a 5% prevalence of small irregular opacities (1/0 or more on the ILO classification) resulted from a cumulative exposure to approximately 1,000 fibre years in Quebec chrysotile miners, to approximately 400 fibre years in Corsican chrysotile miners, and to under 10 fibre years in South African and Australian crocidolite miners. By contrast, for textile workers exposed to Quebec chrysotile, a 5% prevalence of irregular small opacities resulted from a cumulative exposure to under 20 fibre years. Lung dust burden studies are also consistent with a fibre gradient for evoking asbestosis: in 29 men in Pacific shipyard trades with asbestosis associated with mainly amosite exposure, the average lung burden found in autopsy material was 10 million amosite fibres per gram of dry lung tissue compared to an average chrysotile burden of 30 million fibres per gram of dry lung tissue in 23 Quebec chrysotile miners and millers (Becklake and Case 1994). Fibre size distribution contributes to but does not fully explain these differences, suggesting that other plant-specific factors, including other workplace contaminants, may play a role.
Asbestosis may remain stable or progress, but probably does not regress. Progression rates increase with age, with cumulative exposure, and with the extent of existing disease, and are more likely to occur if exposure was to crocidolite. Radiological asbestosis can both progress and appear long after exposure ceases. Deterioration of lung functions may also occur after exposure has ceased (Liddell and Miller 1991). An important issue (and one on which the epidemiological evidence is not consistent) is whether continued exposure increases the chance of progression once radiological changes have developed (Browne 1994; Liddell and Miller 1991). In some jurisdictions, for example in the United Kingdom, the number of cases of asbestosis presenting for worker’s compensation have decreased over the last decades, reflecting the workplace controls put in place in the 1970s (Meredith and McDonald 1994). In other countries, for instance in Germany (Gibbs, Valic and Browne 1994), rates of asbestosis continue to rise. In the United States, age-adjusted asbestos-related mortality rates (based on mention of asbestosis on the death certificate as either the cause of death or as playing a contributory role) for age 1+ increased from under 1 per million in 1960 to over 2.5 in 1986, and to 3 in 1990 (US Dept. of Health and Human Services, 1994).
Diagnosis and case management
Clinical diagnosis depends on:
- establishing the presence of disease
- establishing whether exposure occurred and
- evaluating whether the exposure was likely to have caused the disease.
The chest radiograph remains the key tool to establish the presence of disease, supplemented by HRCT if available in cases where there is doubt. Other objective features are the presence of basal crackles, while lung function level, including exercise challenge, is useful in establishing impairment, a step required for compensation evaluation. Since neither the pathology, radiological changes, nor the symptoms and lung function changes associated with asbestosis are different from those associated with interstitial lung fibrosis due to other causes, establishing exposure is key to diagnosis. In addition, the many uses of asbestos products whose content is often not known to the user makes an exposure history a much more daunting exercise in interrogation than was previously thought. If the exposure history appears inadequate, identification of the agent in biological specimens (sputum, bronchoalveolar lavage and when indicated, biopsy) can corroborate exposure; dose in the form of lung burden can be assessed quantitatively by autopsy or in surgically removed lungs. Evidence of disease activity (from a gallium-67 scan or bronchoalveolar lavage) may assist in estimating prognosis, a key issue in this irreversible condition. Even in the absence of consistent epidemiological evidence that progression is slowed once exposure ceases, such a course may be prudent and certainly desirable. It is not, however, a decision easy to take or recommend, particularly for older workers with little opportunity for job retraining. Certainly exposure should not continue in any workplace not in conformity with current permissible exposure levels. Criteria for the diagnosis of asbestosis for epidemiological purposes are less demanding, particularly for cross-sectional workforce-based studies which include those well enough to be at work. These usually address issues of causality and often use markers that indicate minimal disease, based either on lung function level or on changes in the chest radiograph. By contrast, criteria for diagnosis for medicolegal purposes are considerably more stringent and vary according to the legal administrative systems under which they operate, varying between states within countries as well as between countries.
Asbestos-Related Pleural Disease
Historical perspective
Early descriptions of asbestosis mention fibrosis of the visceral pleura as part of the disease process (see “Pathology”, page 10.55). In the 1930s there were also reports of circumscribed pleural plaques, often calcified, in the parietal pleura (which lines the chest wall and covers the surface of the diaphragm), and occurring in those with environmental, not occupational, exposure. A 1955 workforce-based study of a German factory reported a 5% prevalence of pleural changes on the chest radiograph, thereby drawing attention to the fact that pleural disease might be the primary if not the only manifestation of exposure. Visceroparietal pleural reactions, including diffuse pleural fibrosis, benign pleural effusion (reported first in the 1960s) and rounded atelectasis (first reported in the 1980s) are now all considered interrelated reactions which are usefully distinguished from pleural plaques on the basis of pathology and probably pathogenesis, as well as clinical features and presentation. In jurisdictions in which the prevalence and/or incidence rates of asbestosis are decreasing, pleural manifestations, increasingly common in surveys, are increasingly the basis of detection of past exposure, and increasingly the reason for an individual seeking medical attention.
Pleural plaques
Pleural plaques are smooth, raised, white irregular lesions covered with mesothelium and found on the parietal pleura or diaphragm (figure 6). They vary in size, are often multiple, and tend to calcify with increasing age (Browne 1994). Only a small proportion of those detected at autopsy are seen on the chest radiograph, though most can be detected by HRCT. In the absence of pulmonary fibrosis, pleural plaques may cause no symptoms and may be detected only in screening surveys using chest radiography. Nevertheless, in workforce surveys, they are consistently associated with modest but measurable lung function impairment, mainly in VC and FVC (Ernst and Zejda 1991). In radiological surveys in the United States, rates of 1% are reported in men without known exposure, and 2.3% in men which include those in urban populations, with occupational exposure. Rates are also higher in communities with asbestos industries or high usage rates, while in some workforces, such as sheet metal workers, insulators, plumbers and railroad workers, rates may exceed 50%. In a 1994 Finnish autopsy survey of 288 men aged 35 to 69 years who died suddenly, pleural plaques were detected in 58%, and exhibited the tendency to increase with age, with the probability of exposure (based on history), with the concentration of asbestos fibres in lung tissue, and with smoking (Karjalainen et al. 1994). The aetiologic fraction of plaques attributable to a lung dust burden of 0.1 million fibres per gram of lung tissue was estimated at 24%, (this value is considered to be an underestimate). Lung dust burden studies are also consistent with fibre gradient in potency for evoking pleural reactions; in 103 men with amosite exposure in Pacific shipyard trades, all with pleural plaques, the average autopsy lung burden was 1.4 million fibres per gram of lung tissue, compared to 15.5 and 75 million fibres per gram of lung tissue for chrysotile and tremolite respectively in 63 Quebec chrysotile miners and millers examined in the same way (Becklake and Case 1994).
Figure 6. Asbestos-related pleural disease
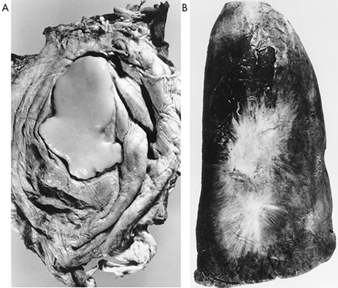
A diaphragmatic pleural plaque (A) is seen in an autopsy specimen as a smooth well defined focus of fibrosis on the diaphragm of a construction worker with incidental exposure to asbestos and asbestos bodies in the lung. Visceral pleural fibrosis (B) is seen on an inflated autopsy lung specimen, and radiates from two central foci on the visceral pleura of the lung of a construction worker with asbestos exposure who also exhibited several parietal pleural plaques. Source: Fraser et al. 1990.
Visceroparietal pleural reactions
Though the pathology and pathogenesis of the different forms of visceroparietal reaction to asbestos exposure are almost certainly interrelated, their clinical manifestations and how they come to attention differs. Acute exudative pleural reactions may occur in the form of effusions in subjects whose lungs do not manifest other asbestos-related disease, or as an exacerbation in the severity and extent of existing pleural reactions. Such pleural effusions are called benign by way of distinguishing them from effusions associated with malignant mesothelioma. Benign pleural effusions occur typically 10 to 15 years after first exposure (or after limited past exposure) in individuals in their 20s and 30s. They are usually transient but may reoccur, may involve one or both sides of the chest simultaneously or sequentially, and may be either silent or associated with symptoms including chest tightness and/or pleural pain and dyspnoea. The pleural fluid contains leucocytes, often blood, and is albumin-rich; only rarely does it contain asbestos bodies or fibres which may, however, be found in biopsy material of the pleura or underlying lung. Most benign pleural effusions clear spontaneously, though in a small proportion of subjects (of the order of 10% in one series) these effusions may evolve into diffuse pleural fibrosis (see figure 6), with or without the development of lung fibrosis. Local pleural reactions may also fold in upon themselves, trapping lung tissue and causing well defined lesions called rounded atelectasis or pseudotumour because they may have the radiological appearance of lung cancer. In contrast to pleural plaques, which seldom cause symptoms, visceroparietal pleural reactions are usually associated with some shortness of breath as well as lung function impairment, particularly when there is obliteration of the costophrenic angle. In one study, for instance, average FVC deficit was 0.07 l when the chest wall was involved and 0.50 l when the costophrenic angle was involved (Ernst and Zejda in Liddell and Miller 1991). As already indicated, the distribution and determinants of pleural reactions vary considerably between workforces, with prevalence rates increasing with:
- estimated residence time of fibre in the lung (measured as time since first exposure)
- exposures primarily to or including amphibole and
- possibly intermittence of exposure, given the high rates of contamination in occupations in which use of asbestos materials is intermittent, but exposure probably heavy.
Lung Cancer
Historical perspective
The 1930s saw the publication of a number of clinical case reports from the United States, the United Kingdom and Germany of lung cancer (a condition much less common then than it is today) in asbestos workers, most of whom also had asbestosis of varying degrees of severity. Further evidence of the association between the two conditions was provided in the 1947 Annual Report of His Majesty’s Chief Inspector of Factories, which noted that lung cancer had been reported in 13.2% of male deaths attributed to asbestosis in the period 1924 to 1946 and in only 1.3% of male deaths attributed to silicosis. The first study to address the causal hypothesis was a cohort mortality study of a large United Kingdom asbestos textile plant (Doll 1955), one of the first such workforce-based studies, and by 1980, after at least eight such studies in as many workforces had confirmed an exposure-response relationship, the association was generally accepted as causal (McDonald and McDonald in Antman and Aisner 1987).
Clinical features and pathology
In the absence of other associated asbestos disease, the clinical features and criteria for the diagnosis of asbestos-associated lung cancer are no different from those for lung cancer not associated with asbestos exposure. Originally, asbestos-associated lung cancers were considered to be scar cancers, similar to lung cancer seen in other forms of diffuse lung fibrosis such as scleroderma. Features which favoured this view were their location in the lower lung lobes (where asbestosis is usually more marked), their sometimes multicentric origin and a preponderance of adenocarcinoma in some series. However, in most reported workforce-based studies, the distribution of cell types was no different from that seen in studies of non-asbestos-exposed populations, supporting the view that asbestos itself may be a human carcinogen, a conclusion reached by the International Agency for Research on Cancer (World Health Organization: International Agency for Research on Cancer 1982). Most but not all asbestos-related lung cancers occur in association with radiologic asbestosis (see below).
Epidemiology
Cohort studies confirm that lung cancer risk increases with exposure, though the fractional rate of increase for each fiber per milliliter per year exposed varies, and is related both to fibre type and to industrial process (Health Effects Institute—Asbestos Research 1991). For instance, for mainly chrysotile exposures in mining, milling and friction product manufacture, the increase ranged from approximately 0.01 to 0.17%, and in textile manufacture from 1.1 to 2.8%, while for exposure to amosite insulation products and some cement product exposures involving mixed fibre, rates of as high as 4.3 and 6.7% have been recorded (Nicholson 1991). Cohort studies in asbestos workers also confirm that cancer risk is demonstrable for non-smokers and that risk is increased (closer to multiplicative than additive) by cigarette smoking (McDonald and McDonald in Antman and Aisner 1987). The relative risk for lung cancer declines after exposure ceases, although the decline appears slower than that which occurs after quitting smoking. Lung dust burden studies are also consistent with a fibre gradient in lung cancer production; 32 men in Pacific shipyard trades with mainly amosite exposure had a lung dust burden of 1.1 million amosite fibres per gram of dry lung tissue compared to 36 Quebec chrysotile miners with an average lung dust burden of 13 million chrysotile fibres per gram of lung tissue (Becklake and Case 1994).
Relationship to asbestosis
In the 1955 autopsy study of causes of death in 102 workers employed in the United Kingdom asbestos textile factory referred to above (Doll 1955), lung cancer was found in 18 individuals, 15 of whom also had asbestosis. All subjects in whom both conditions were found had worked for at least 9 years before 1931, when national regulations for asbestos dust control were introduced. These observations suggested that as exposure levels decreased, the competing risk of death from asbestosis also decreased and workers lived long enough to exhibit the development of cancer. In most workforce-based studies, older workers with long service have some pathological evidence of asbestosis (or asbestos-related small airways disease) at autopsy even though this may be minimal and not detectable on the chest radiograph in life (McDonald and McDonald in Antman and Aisner 1987). Several but not all cohort studies are consistent with the view that not all excess lung cancers in populations exposed to asbestos are related to asbestosis. More than one pathogenetic mechanism may in fact be responsible for lung cancers in individuals exposed to asbestos depending on the site and deposition of the fibres. For instance, long thin fibres, which are deposited preferentially at airway bifurcations, are thought to become concentrated and to act as inducers of the process of cancerogenesis through chromosomal damage. Promoters of this process may include continued exposure to asbestos fibres or to tobacco smoke (Lippman 1995). Such cancers are more likely to be squamous cell in type. By contrast, in lungs which are the site of fibrosis, cancerogenesis may result from the fibrotic process: such cancers are more likely to be adenocarcinomas.
Implications and attributability
While determinants of excess cancer risk can be derived for exposed populations, attributability in the individual case cannot. Obviously, attributability to asbestos exposure is more likely and credible in an exposed individual with asbestosis who has never smoked than in an exposed individual without asbestosis who smokes. Nor can this probability be modelled reasonably. Lung dust burden measurements may supplement a careful clinical assessment but each case must be evaluated on its merits (Becklake 1994).
Malignant Mesothelioma
Pathology, diagnosis, ascertainment and clinical features
Malignant mesotheliomas arise from the serous cavities of the body. Approximately two-thirds arise in the pleura, about one-fifth in the peritoneum, while the pericardium and tunica vaginalis are much less frequently affected (McDonald and McDonald in Lidell and Miller 1991). Since mesothelial cells are pluripotential, the histological features of mesothelial tumours may vary; in most series, epithelial, sarcomatous and mixed forms account for approximately 50, 30 and 10% of cases respectively. Diagnosis of this rare tumour, even in the hands of experienced pathologists, is not easy, and mesothelioma panel pathologists often confirm only a small percentage, in some studies less than 50% of cases submitted for review. A variety of cytological and immunohistochemical techniques have been developed to assist in differentiating malignant mesothelioma from the main alternative clinical diagnoses, namely, secondary cancer or reactive mesothelial hyperplasia; this remains an active research field in which expectations are high but findings inconclusive (Jaurand, Bignon and Brochard 1993). For all these reasons, ascertainment of cases for epidemiological surveys is not straightforward, and even when based on cancer registries, may be incomplete. In addition, confirmation by expert panels using specified pathological criteria is necessary to assure comparability in criteria for registration.
Clinical features
Pain is usually the presenting feature. For pleural tumours, this starts in the chest and/or shoulders, and may be severe. Breathlessness follows, associated with pleural effusion and/or progressive encasement of the lung by tumour, and weight loss. With peritoneal tumours, abdominal pain is usually accompanied by swelling. Imaging features are illustrated in figure 7. The clinical course is usually rapid and median survival times, six months in a 1973 report and eight months in a 1993 report, have changed little over the last two decades, despite the greater public and medical awareness which often leads to earlier diagnosis and despite advances in diagnostic techniques and an increase in the number of treatment options for cancer.
Figure 7. Malignant mesothelioma
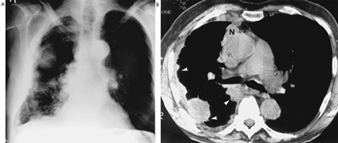
Seen on an overpenetrated chest roetngenogram (A) as a large mass in the axillary region. Note the associated reduction in volume of the right haemothorax with marked irregular nodular thickening of the pleura of the whole right lung. CT scan (B) confirms the extensive pleural thickening involving parietal and mediastinal pleura (closed arrows) in and around the ribs. Source: Fraser et al. 1990
Epidemiology
In the 15 years which followed the 1960 report of the mesothelioma case series from the Northwest Cape, South Africa (Wagner 1996), international confirmation of the association came from reports of other case series from Europe (United Kingdom, France, Germany, Holland), the United States (Illinois, Pennsylvania and New Jersey) and Australia, and of case control studies from the United Kingdom (4 cities), Europe (Italy, Sweden, Holland) and from the United States and Canada. Odds ratios in these studies ranged from 2 to 9. In Europe in particular, the association with shipyard occupations was strong. In addition, proportional mortality studies in asbestos-exposed cohorts suggested that risk was associated both with fibre type and with industrial process, with rates attributable to mesothelioma ranging from 0.3% in chrysotile mining to 1% in chrysotile manufacturing, compared with 3.4% in amphibole mining and manufacturing and as high as 8.6% for exposure to mixed fibre in insulation (McDonald and McDonald in Liddell and Miller 1991). Similar fibre gradients are shown in cohort mortality studies which, given the short survival times of these tumours, are a reasonable reflection of incidence. These studies also show longer latent periods when exposure was to chrysotile compared to amphiboles. Geographical variation in incidence has been documented using Canadian age-and sex-specific rates for 1966 to 1972 to calculate expected rates (McDonald and McDonald in Liddell and Miller 1991); rate ratios (values actually observed over expected) were 0.8 for the United States (1972), 1.1 for Sweden (1958 to 1967), 1.3 for Finland (1965 to 1969), 1.7 for United Kingdom (1967 to 1968), and 2.1 for the Netherlands (1969 to 1971). While technical factors including ascertainment may obviously contribute to the variation recorded, the results do suggest higher rates in Europe than in North America.
Time trends and gender differences in mesothelioma incidence have been used as a measure of the health impact of asbestos exposure on populations. The best estimates for overall rates in industrialized countries before 1950 are under 1.0 per million for men and women (McDonald and McDonald in Jaurand and Bignon 1993). Subsequently, rates increased steadily in men and either not at all or less in women. For instance, overall rates in men and women per million were reported at 11.0 and under 2.0 in the United States in 1982, 14.7 and 7.0 in Denmark for 1975-80, 15.3 and 3.2 in the United Kingdom for 1980-83, and 20.9 and 3.6 in the Netherlands for 1978-87. Higher rates in men and women, but excluding younger subjects, were reported for crocidolite mining countries: 28.9 and 4.7 respectively in Australia (aged 2+) for 1986, and 32.9 and 8.9 respectively in South African Whites (aged 1+) for 1988 (Health Effects Institute—Asbestos Research 1991). The rising rates in men are likely to reflect occupational exposure, and if so, they should level off or decrease within the 20-to 30-year “incubation” period following the introduction of workplace controls and reduction of exposure levels in most workplaces in most industrialized countries in the 1970s. In countries in which the rates in women are rising, this increase may reflect their increasing engagement in occupations with risk exposure, or the increasing environmental or indoor contamination of urban air (McDonald 1985).
Aetiology
Environmental factors are clearly the main determinants of mesothelioma risk, exposure to asbestos being the most important, though the occurrence of family clusters maintains interest in the potential role of genetic factors. All asbestos fibre types have been implicated in mesothelioma production, including anthophyllite for the first time in a recent report from Finland (Meurman, Pukkala and Hakama 1994). However, there is a substantial body of evidence, from proportional and cohort mortality studies and lung burden studies, which suggests the role of a fibre gradient in mesothelioma production, risk being higher for exposures to mainly amphiboles or amphibole chrysotile mixtures, compared with mainly chrysotile exposures. In addition, there are rate differences between workforces for the same fibre at what appears to be the same exposure level; these remain to be explained, though fibre size distribution is a likely contributing factor.
The role of tremolite has been widely debated, a debate sparked by the evidence of its biopersistence in lung tissue, animal and human, compared to that of chrysotile. A plausible hypothesis is that the many short fibres which reach and are deposited in peripheral lung airways and alveoli are cleared to subpleural lymphatics where they collect; their potency in mesothelioma production depends on their biopersistence in contact with pleural surfaces (Lippmann 1995). In human studies, mesothelioma rates are lower for populations exposed at work to chrysotile relatively uncontaminated by tremolite (for instance, in Zimbabwean mines) compared to those exposed to chrysotile which is so contaminated (for instance, in Quebec mines), and these findings have been replicated in animal studies (Lippmann 1995). Also, in a multivariate analysis of lung fibre burden in material from a Canada-wide mesothelioma case control study (McDonald et al. 1989), the results suggested that most if not all mesotheliomas could be explained by tremolite lung fibre burden. Finally, a recent analysis of the mortality in the cohort of over 10,000 Quebec chrysotile miners and millers born between 1890 and 1920, and followed to 1988 (McDonald and McDonald 1995), supports this view: in almost 7,300 deaths, the 37 mesothelioma deaths were concentrated in certain mines from the Thetford area, yet the lung burden of 88 cohort members from the mines implicated did not differ from that of miners from other mines in terms of chrysotile fibre burden, only in terms of tremolite burden (McDonald et al. 1993).
What has been called the tremolite question is perhaps the most important of the currently debated scientific issues, and it also has public health implications. Note must also be made of the important fact that in all series and jurisdictions, a certain proportion of cases occur without reported asbestos exposure, and that only in some of these cases do lung dust burden studies point to previous environmental or occupational exposure. Other occupational exposures have been implicated in mesothelioma production, for instance in talc, vermiculite and possibly mica mining, but in these, the ore contained either tremolite or other fibres (Bignon, Peto and Saracci 1989). An open search for other exposures, occupational or non-occupational, to fibres, inorganic and organic, and to other agents which may be associated with mesothelioma production, should continue.
Other Asbestos-Related Diseases
Chronic airways disease
Usually included under this rubric are chronic bronchitis and chronic obstructive pulmonary disease (COPD), both of which can be diagnosed clinically, and emphysema, until recently diagnosed only by pathological examination of lungs removed at autopsy or otherwise (Becklake 1992). A major cause is smoking, and, over the past decades, mortality and morbidity due to chronic airways disease has increased in most industrialized countries. However, with the decline of pneumoconiosis in many workforces, evidence has emerged to implicate occupational exposures in the genesis of chronic airways disease, after taking into account the dominant role of smoking. All forms of chronic airways disease have been shown to be associated with work in a variety of dusty occupations, including those occupations in which an important component of the dust contaminating the workplace was asbestos (Ernst and Zejda in Liddell and Miller 1991). Total pollutant burden, rather than exposure to any of its particular components, in this case asbestos dust, is thought to be implicated, in much the same way as the effect of smoking exposure on chronic airways diseases is viewed, that is, in terms of total exposure burden (e.g., as pack-years), not exposure to any one of the over 4,000 constituents of tobacco smoke. (see elsewhere in this volume for a further discussion of the relationship between occupational exposures and chronic airways disease).
Other cancers
In several of the earlier cohort studies of asbestos exposed workers, mortality attributable to all cancers exceeded that expected, based on national or regional vital statistics. While lung cancer accounted for most of the excess, other cancers implicated were gastro-intestinal cancers, laryngeal cancer and cancer of the ovaries, in that order of frequency. For gastro-intestinal cancers, (including those affecting the oesophagus, the stomach, the colon and the rectum), the relevant exposure in occupational cohorts is presumed to be via swallowing asbestos-laden sputum raised from the major airways in the lung, and in earlier times, (before protection measures were taken against exposure at lunch sites) direct contamination of food in workplaces which had no lunch areas separate from working areas of plants and factories. Retrograde flow via the thoracic duct from lymph nodes draining the lung might also occur (see “Fate of inhaled fibres”, page 10.54). Because the association was inconsistent in the different cohorts studied, and because exposure response relationships were not always seen, there has been a reluctance to accept the evidence of the association between occupational exposure and asbestos exposure as causal (Doll and Peto 1987; Liddell and Miller 1991).
Cancer of the larynx is much less common than gastro-intestinal or lung cancer. As early as the 1970s, there were reports of an association between cancer of the larynx and asbestos exposure. Like lung cancer, a major risk factor and cause of laryngeal cancer is smoking. Laryngeal cancer is also strongly associated with alcohol consumption. Given the location of the larynx (an organ exposed to all the inhaled pollutants to which the lungs are exposed) and given the fact that it is lined by the same epithelium that lines the major bronchi, it is certainly biologically plausible that cancer of the larynx occurs as a result of asbestos exposure. However, the overall evidence available to date is inconsistent, even from large cohort studies such as the Quebec and Balangero (Italy) chrysotile miners, possibly because it is a rare cancer and there is still reluctance to regard the association as causal (Liddell and Miller 1991) despite its biological plausibility. Cancer of the ovaries has been recorded in excess of expected in three cohort studies (WHO 1989). Misdiagnosis, in particular as peritoneal mesothelioma, may explain most of the cases (Doll and Peto 1987).
Prevention, Surveillance and Assessment
Historical and current approaches
Prevention of any pneumoconiosis, including asbestosis, has traditionally been through:
- engineering and work practices to maintain airborne fibre levels as low as possible, or at least in conformity with permissible exposure levels usually set by law or regulation
- surveillance, conducted to record trends of markers of disease in exposed populations and monitor the results of control measures
- education and product labelling aimed at assisting workers as well as the general public in avoiding non-occupational exposure.
Permissible exposure levels were originally directed at controlling asbestosis and were based on industrial hygiene measurements in million particles per cubic foot, gathered using the same methods as were used for the control of silicosis. With the shift in biological focus to fibres, in particular long thin ones, as the cause of asbestosis, methods more appropriate to their identification and measurement in air were developed and, given these methods, the focus on the more abundant short fibres which contaminate most workplaces was minimized. Aspect (length to diameter) ratios for most particles of milled chrysotile asbestos fall within the range 5:1 to 20:1, going up to 50:1, in contrast to most particles of milled amphibole asbestos (including cleavage fragments) whose values fall below 3:1. The introduction of the membrane filter for fibre counting of air samples led to an arbitrary industrial hygiene and medical definition of a fibre as a particle at least 5μm long, 3μm or less thick, and with a length to width ratio of at least 3:1. This definition, used for many of the studies of exposure-response relationships, forms the scientific basis for setting environmental standards.
For instance, it was used in a meeting sponsored by the World Health Organization (1989) to propose occupational exposure limits and has been adopted by agencies such as the US Occupational Safety and Health Administration; it is retained mainly for reasons of comparability. The WHO meeting, chaired by Sir Richard Doll, while recognizing that the occupational exposure limit in any country can only be set by the appropriate national body, recommended that countries having high limits should take urgent steps to lower the occupational exposure for an individual worker to 2 f/ml (eight-hour time-weighted average) and that all countries should move as quickly as possible to 1 f/ml (eight-hour time-weighted average) if they had not already done so. With the decrease in asbestosis rates in some industrialized countries, and concern over asbestos-related cancers in all, attention has now shifted to determining whether the same fibre parameters—that is, at least 5mm long, 3mm or less thick, and with a length to width ratio of at least 3:1—are also appropriate for controlling carcinogenesis (Browne 1994). A current theory of asbestos carcinogenesis implicates short as well as long fibres (Lippmann 1995). In addition, given the evidence for a fibre gradient in mesothelioma and lung cancer production, and to a lesser extent, for asbestosis production, an argument could be made for permissible exposure levels taking fibre type into account. Some countries have addressed the issue by banning the use (and thus the import) of crocidolite, and setting more stringent exposure levels for amosite, namely 0.1 f/l (McDonald and McDonald 1987).
Exposure levels in the workplace
Permissible exposure levels embody the hypothesis, based on all available evidence, that human health will be preserved if exposure is maintained within those limits. Revision of permissible exposure levels, when it occurs, is invariably towards greater stringency (as described in the paragraph above). Nevertheless, despite good compliance with workplace controls, cases of disease continue to occur, for reasons of personal susceptibility (for instance, higher-than-average fibre retention rates) or because of failure of workplace controls for certain jobs or processes. Engineering controls, improved workplace practices and the use of substitutes, described elsewhere in the chapter, have been implemented internationally (Gibbs, Valic and Browne 1994) in larger establishments through industry, union and other initiatives. For instance, according to a 1986 worldwide industry review, compliance with the current recommended standard of 1 f/ml had been achieved at 83% of production sites (mines and mills) covering 13,499 workers in 6 countries; in 96% of 167 cement factories operating in 23 countries; in 71% of 40 textile factories covering over 2,000 workers operating in 7 countries; and in 97% of 64 factories manufacturing friction materials, covering 10,190 workers in 10 countries (Bouige 1990). However, a not unimportant proportion of such workplaces still do not comply with regulations, not all manufacturing countries participated in this survey, and the anticipated health benefits are evident only in some national statistics, not in others (“Diagnosis and case management”, page 10.57). Control in demolition processes and small enterprises using asbestos continues to be less than successful, even in many industrialized countries.
Surveillance
The chest radiograph is the main tool for asbestosis surveillance, cancer registries and national statistics for asbestos-related cancers. A commendable initiative in international surveillance of mining, tunnelling and quarrying, undertaken by the ILO through voluntary reporting from governmental sources, focuses on coal and hard-rock mining but could include asbestos. Unfortunately, follow-through has been poor, with the last report, which was based on data for 1973-77, being published in 1985 (ILO 1985). Several countries issue national mortality and morbidity data, an excellent example being the Work-related Lung Disease Surveillance Report for the United States, a report referred to above (USDHSS 1994). Such reports provide information to interpret trends and evaluate the impact of control levels at a national level. Larger industries should (and many do) keep their own surveillance statistics, as do some unions. Surveillance of smaller industries may require specific studies at appropriate intervals. Other sources of information include programmes such as the Surveillance of Work-related Respiratory Diseases (SWORD) in the United Kingdom, which gathers regular reports from a sample of the country’s chest and occupational physicians (Meredith and McDonald 1994), and reports from compensation boards (which often, however, do not provide information on workers at risk).
Product labelling, education and the information highway
Mandatory product labelling together with worker education and education of the general public are powerful tools in prevention. While in the past, this took place within the context of worker organizations, worker management committees, and union education programmes, future approaches could exploit electronic highways to make available databases on health and safety in toxicology and medicine.
Exposure in buildings and from water supplies
In 1988, a review of potential health risks associated with working in buildings constructed using asbestos-containing materials was mandated by the US Congress (Health Effects Institute—Asbestos Research 1991). The results of a large number of indoor sampling studies from Europe, the United States and Canada were used in risk estimates. The lifetime risk for premature cancer death was estimated to be 1 per million for those exposed for 15 years in schools (for estimated exposure levels ranging from .0005 to .005 f/ml) and 4 per million for those exposed for 20 years working in office buildings (for estimated exposure levels ranging from .0002 to .002 f/ml). For comparison, the risk for occupational exposure to 0.1 f/ml (i.e., in compliance with the permissible exposure limit proposed by the US Occupational Safety and Health Administration) for 20 years was estimated at 2,000 per million exposed. Measurements in drinking water in urban communities show considerable variation, from undetectable levels to high levels ranging from 0.7 million f/l in Connecticut, USA, to levels ranging from 1.1 million to 1.3 billion f/l in the mining areas of Quebec (Bignon, Peto and Saracci 1989). Some contamination may also occur from the asbestos cement pipes which must service most urban water reticulation services in the world. However, a working group which reviewed the evidence in 1987 did not discount the potential associated hazard, but did not regard the health risks associated with asbestos ingestion as “one of the most pressing public health hazards” (USDHHS 1987), a view concordant with the concluding remarks in an IARC (WHO) monograph on non-occupational exposure to mineral fibres (Bignon, Peto and Saracci 1989).
Asbestos and other fibres in the 21st century
The first half of the twentieth century was characterized by what could be described as gross neglect of asbestos-related ill health. Before the Second World War, the reasons for this are not clear; the scientific basis for control was there but perhaps not the will and not the worker militancy. During the war, there were other national and international priorities, and after the war, pressures of urbanization by a rapidly increasing world population took precedence, and perhaps fascination in an industrial age with the versatility of the “magic” mineral diverted attention from its dangers. Following the first International Conference on the Biological Effects of Asbestos in 1964 (Selikoff and Churg 1965), asbestos-related disease became a cause célèbre, not only on its own account, but also because it marked a period of labour-management confrontation concerning the rights of the worker to knowledge about workplace hazards, health protection and fair compensation for injury or illness. In countries with no-fault worker’s compensation, asbestos-related disease on the whole received fair recognition and handling. In countries where product liability and class action suits were more usual, large awards have been made to some affected workers (and their lawyers) while others have been left destitute and without support. While the need for fibres in modern societies is unlikely to diminish, the role of the mineral fibres vis-à-vis other fibres may change. There has already been a shift in uses both within and between countries (see “Other sources of exposure”, page 10.53). Though the technology exists to diminish workplace exposures, there remain workplaces in which it has not been applied. Given the current knowledge, given international communication and product labelling, and given worker education and industry commitment, it should be possible to use this mineral to provide cheap and durable products for use in construction and water reticulation on an international basis without risk to user, worker, manufacturer or miner, or to the general public at large.
Hard Metal Disease
Shortly after the end of the First World War, while doing research to find a material able to replace diamond in metal-drawing nozzles, Karl Schoeter patented in Berlin a sintering process (pressurization plus heating at 1,500°C) of a mixture of fine tungsten carbide (WC) powder with 10% of cobalt to produce “hard metal”. The main characteristics of this sinter are the extreme hardness, only slightly inferior to that of diamond, and the maintenance of its mechanical properties at high temperatures; these characteristics make it suitable for use in drawing metal, for welded inserts, and for high-speed tools for machining of metals, stone, wood and materials with high resistance to wear or to heat, in the mechanical, aeronautical and ballistic fields. The use of hard metal is continually expanding all over the world. In 1927 Krupp extended the use of hard metal into the cutting tools field, calling it “Widia” (wie Diamant—like diamond), a name still in use today.
Sintering remains the basis of all hard metal production: techniques are improved by the introduction of other metallic carbides—titanium carbide (TiC) and tantalum carbide (TaC)—and by treatment of hard metal parts for mobile cutting inserts with one or more layers of titanium nitride or aluminium oxide and of other very hard compounds applied with chemical vapour deposition (CVD) or physical vapour deposition (PVD). The fixed inserts welded to the tools cannot be plated, but are repeatedly sharpened by a diamond grinding wheel (figures 1 and 2).
Figure 1. (A) Examples of some hard metal drawing mobile inserts, plated with golden-yellow tungsten nitride; (B) insert welded to the tool and working in steel drawing.

Figure 2. Fixed inserts welded to (A) stone drill and (B) saw disk.

The hard metal sinter is formed by particles of metallic carbides incorporated in a matrix formed by cobalt, which melts during sintering, interacting and occupying the interstices. Cobalt is therefore the structure gluing material, which assumes metal-ceramic characteristics (figures 3, 4, and 5).
Figure 3. Microstructure of a WC/Co sintering; WC particles are incorporated into the Co light matrix (1,500x).

Figure 4. Microstructure of a WC + TiC + TaC + Co sintering. Along with WC prismatic particles, globular particles formed by a solid solution of TiC + TaC are observed. The light matrix is formed by Co (1,500x).

Figure 5. Sintering microstructure plated by multiple very hard layers (2,000x).
The sintering process uses very fine metallic carbide powders (average diameters from 1 to 9μm) and cobalt powders (average diameter from 1 to 4μm) which are mixed, treated with paraffin solution, die-pressed, de-waxed at low temperature, pre-sintered at 700 to 750°C and sintered at 1,500°C (Brookes 1992).
When sintering is done with inadequate methods, improper techniques and poor industrial hygiene, the powders can pollute the atmosphere of the work environment: workers are therefore exposed to the risk of inhalation of metallic carbide powders and cobalt powders. Along with the primary process there are other activities which can expose the workers to the risk of aerosol inhalation of hard metal. Sharpening of fixed inserts welded to tools is normally carried out by dry diamond grinding or, more frequently, cooled with liquids of different kinds, producing powders or mists formed by very small drops containing metallic particles. Particles of hard metal are also used in the production of a high-resistance layer on steel surfaces subjected to wear, applied through methods (plasma coating process and others) based on the combination of a powder spray with an electric arc or a controlled explosion of a gas mixture at high temperature. The electric arc or the explosive flow of the gas determines the fusion of the metallic particles and their impact on the surface being plated.
First observations on “hard metal diseases” were described in Germany in the 1940s. They reported a diffuse, progressive pulmonary fibrosis, called Hartmetallungenfibrose. During the next 20 years parallel cases were observed and described in all industrial countries. The workers affected were in the majority of cases in charge of the sintering. From 1970 to the present, several studies indicate that the pathology to the breathing apparatus is caused by the inhalation of hard metal particles. It affects only susceptible subjects, and consists of the following symptoms:
- acute: rhinitis, asthma
- subacute: fibrosant alveolitis
- chronic: diffuse and progressive interstitial fibrosis.
It affects not only workers in charge of sintering, but anyone inhaling aerosol containing hard metal and particularly cobalt. It is mainly and perhaps exclusively caused by cobalt.
The definition of hard metal disease now includes a group of pathologies of the breathing apparatus, different from each other in clinical gravity and prognosis, but having in common a variable individual reactivity to the aetiological factor, cobalt.
More recent epidemiological and experimental information agree on the causal role of cobalt for acute symptoms in the upper respiratory tract (rhinitis, asthma) and for subacute and chronic symptoms in the bronchial parenchyma (fibrosing alveolitis and chronic interstitial fibrosis).
The pathogenic mechanism is based on the induction by Co of a hypersensitive immunoreaction: in fact, only some of the subjects present pathologies after short exposures to relatively low concentration, or even after longer and more intense exposures. Co concentrations in biological samples (blood, urine, skin) are not significantly different in those who have the pathology and those who do not; there is no correlation of dose and response at the tissue level; specific antibodies have been individuated (immunoglobins IgE and IgG) against a Co-albumin compound in asthmatics, and the Co patch test is positive in the subjects with alveolitis or fibrosis; the cytological aspects of the giant-cellular alveolitis are compatible with immunoreaction, and acute or subacute symptoms tend to regress when the subjects are removed from exposure to Co (Parkes 1994).
The immunological basis of hypersensitivity to Co has not yet been satisfactorily explained; it is not possible, therefore, to identify a reliable marker of individual susceptibility.
Identical pathologies to those found in the subjects exposed to hard metals were also observed in diamond cutters, who use disks formed by microdiamonds cemented with Co and who therefore inhale only Co and diamond particles.
It is not yet fully demonstrated that pure Co (all other inhaled particles excluded) is capable alone of producing the pathologies and above all the diffused interstitial fibrosis: the particles inhaled with Co could have a synergistic as well as modulating effect. Experimental studies seem to demonstrate that the biological reactivity to a mixture of Co particles and of tungsten is stronger than that caused by Co alone, and significant pathologies are not to be observed in the workers in charge of the production of pure Co powder (Science of the Total Environment 1994).
Clinical symptoms of hard metal disease, which, on the basis of current aetiopathogenic knowledge should be more precisely called “cobalt disease”, are, as mentioned before, acute, subacute and chronic.
Acute symptoms include a specific respiratory irritation (rhinitis, laryngo-tracheitis, pulmonary oedema) caused by exposure to high concentrations of Co powder or Co smoke; they are observable only in exceptional cases. Asthma is observed more frequently. It appears in 5 to 10% of the workers exposed to cobalt concentrations of 0.05 mg/m3, the current US threshold limit value (TLV). Symptoms of thoracic constriction with dyspnoea and cough tend to appear at the end of the work shift or during the night. The diagnosis of occupational allergic bronchial asthma due to cobalt can be suspected on the basis of case history criteria, but it is confirmed by a specific bronchial stimulation test which determines the appearance of an immediate, delayed or dual bronchospastic response. Even respiratory capacity tests carried out at the beginning and at the end of the work shift can help the diagnosis. Asthmatic symptoms due to cobalt tend to disappear when the subject is removed from exposure, but, similarly to all other forms of occupational allergic asthma, symptoms can become chronic and irreversible when the exposure continues for a long time (years), despite the presence of respiratory disturbances. Highly bronchoreactive subjects can present non-allergic aetiological asthmatic symptoms, with a non-specific response to inhalation of cobalt and other irritating powders. In a high percentage of cases with allergic bronchial asthma, specific reaction towards a human Co-seroalbumin compound was found in the IgE serum. The radiological finding does not vary: only in rare cases can mixed forms of asthma plus alveolitis with radiological alteration specifically caused by alveolitis be found. Bronchodilator therapy, along with an immediate end of the work exposure, leads to complete recovery for cases that are of recent onset, not yet chronic.
Subacute and chronic symptoms include fibrosant alveolitis and chronic diffuse and progressive interstitial fibrosis (DIPF). The clinical experience seems to indicate that the transition from alveolitis to interstitial fibrosis is a process which evolves gradually and slowly in time: one can find cases of pure initial alveolitis reversible with withdrawal from the exposure plus corticosteroid therapy; or cases with an already present fibrosis component, which can improve but not reach complete recovery by removing the subject from exposure, even with additional therapy; and finally, cases in which the predominant situation is that of an irreversible DIPF. The occurrence of such cases is low in the exposed workers, very much lower than the percentage of allergic asthma cases.
Alveolitis is easy to study today in its cytological components through broncho-alveolar lavage (BAL); it is characterized by a large increase of the total cell number, mainly formed by macrophages, with numerous multinuclear giant cells and the typical aspect of foreign-body giant cells containing at times cytoplasmic cells (figure 6); even an absolute or relative increase of lymphocytes is frequent, with a decreased CD4/CD8 ratio, associated with a large increase of eosinophiles and mast cells. Rarely, alveolitis is mainly lymphocytic, with CD4/CD8 ratio inverted, as it occurs in the pneumopathies due to hypersensitivity.
Figure 6. Cytological BAL in a macrophagic mono-nuclear giant-cellular alveolitis case caused by hard metal. Between the mononuclear macrophages and the lymphocyte, a giant foreign-body type of cell (400x) is observed.
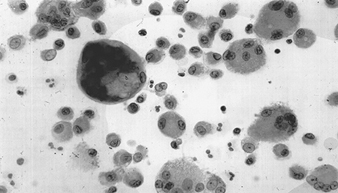
Subjects with alveolitis report dyspnoea linked with fatigue, loss of weight and dry cough. Crepitation is present in the lower lung with functional alteration of a restrictive kind and diffused round or irregular radiological opacity. The patch test for cobalt is positive in the majority of cases. In the susceptible subjects, alveolitis is revealed after a relatively short period of workplace exposure, of one or a few years. In its initial phases this form is reversible up to complete recovery with the simple removal from exposure, with better results if this is combined with cortisone therapy.
The development of diffuse interstitial fibrosis aggravates the clinical symptoms with a worsening of the dyspnoea, which appears even after minimal strain and then even at rest, with worsening of the restrictive ventilatory impairment which is linked to a reduction of the capillary-alveolar diffusion, and with appearance of radiographic opacities of a linear type and of honeycombing (figure 7). The histological situation is that of a fibrosing alveolitis of a “mural type”.
Figure 7. Thoracic radiograph of a subject affected by interstitial fibrosis caused by hard metal. Linear and diffused opacity and honeycombing aspects are observed.
The evolution is rapidly progressive; therapies are ineffective and the prognosis doubtful. One of the cases diagnosed by the author eventually required a lung transplant.
The occupational diagnosis is based on case history, BAL cytological pattern and cobalt patch test.
Prevention of hard metal disease, or, more precisely, of cobalt disease, is now mainly technical: protecting the workers through the elimination of powder, smokes or mists with adequate ventilation of the work areas. In fact, the lack of knowledge about the factors which determine individual hypersensitivity to cobalt makes the identification of susceptible people impossible, and the maximum effort must be made to reduce the atmospheric concentrations.
The number of people at risk is underestimated because many sharpening activities are carried out in small industries or by craftspeople. In such workplaces, the US TLV of 0.05 mg/m3 is frequently exceeded. There is also some question as to the adequacy of the TLV for protecting workers against cobalt disease since dose-effect relationships for disease mechanisms involving hypersensitivity are not completely understood.
Routine surveillance must be accurate enough to identify cobalt pathologies in their earliest stages. An annual questionnaire aimed mainly at temporary symptoms must be administered, along with a medical examination that includes pulmonary function testing and other appropriate medical examinations. Since it has been demonstrated that there is a good correlation between cobalt concentrations in the work environment and the urinary excretion of the metal, it is appropriate to carry out semi-annual measurement of cobalt in urine (CoU) on samples taken at the end of the work week. When the exposure is at the level of the TLV, the biological exposure index (BEI) is estimated to be equal to 30μg Co/litre urine.
Pre-exposure medical examinations for the presence of pre-existing respiratory disease and bronchial hypersensitivity can be useful in the counselling and placement of workers. Metacholine tests are a useful indicator of non-specific bronchial hyperreactivity and may be useful in some settings.
International standardization of environmental and medical surveillance methods for workers exposed to cobalt is highly recommended.
Respiratory System: The Variety of Pneumoconioses
This article is devoted to a discussion of pneumoconioses related to a variety of specific non-fibrous substances; exposures to these dusts are not covered elsewhere in this volume. For each material capable of engendering a pneumoconiosis upon exposure, a brief discussion of the mineralogy and commercial importance is followed by information related to the lung health of exposed workers.
Aluminium
Aluminium is a light metal with many commercial uses in both its metallic and combined states. (Abramson et al. 1989; Kilburn and Warshaw 1992; Kongerud et al. 1994.) Aluminium-containing ores, primarily bauxite and cryolite, consist of combinations of the metal with oxygen, fluorine and iron. Silica contamination of the ores is common. Alumina (Al2O3) is extracted from bauxite, and may be processed for use as an abrasive or as a catalyst. Metallic aluminium is obtained from alumina by electrolytic reduction in the presence of fluoride. Electrolysis of the mixture is carried out by using carbon electrodes at a temperature of about 1,000°C in cells known as pots. The metallic aluminium is then drawn off for casting. Dust, fume and gas exposures in pot rooms, including carbon, alumina, fluorides, sulphur dioxide, carbon monoxide and aromatic hydrocarbons, are accentuated during crust breaking and other maintenance operations. Numerous products are manufactured from aluminium plate, flake, granules and castings—resulting in extensive potential for occupational exposures. Metallic aluminium and its alloys find use in the aircraft, boat and automobile industries, in the manufacture of containers and of electrical and mechanical devices, as well as in a variety of construction and structural applications. Small aluminium particles are used in paints, explosives and incendiary devices. To maintain particle separation, mineral oils or stearin are added; increased lung toxicity of aluminium flakes has been associated with the use of mineral oil.
Lung health
Inhalation of aluminium-containing dusts and fumes may occur in workers involved in the mining, extraction, processing, fabrication and end-use of aluminium-containing materials. Pulmonary fibrosis, resulting in symptoms and radiographic findings, has been described in workers with several differing exposures to aluminium-containing substances. Shaver’s disease is a severe pneumoconiosis described among workers involved in the manufacture of alumina abrasives. A number of deaths from the condition have been reported. The upper lobes of the lung are most often affected and the occurrence of pneumothorax is a frequent complication. High levels of silicon dioxide have been found in the pot room environment as well as in workers’ lungs at autopsy, suggesting silica as a potential contributor to the clinical picture in Shaver’s disease. High concentrations of aluminium oxide particulate have also been observed. Lung pathology may show blebs and bullae, and pleural thickening is seen occasionally. The fibrosis is diffuse, with areas of inflammation in the lungs and associated lymph nodes.
Aluminium powders are used in making explosives, and there have been a number of reports of a severe and progressive fibrosis in workers involved in this process. Lung involvement has also occasionally been described in workers employed in the welding or polishing of aluminium, and in bagging cat litter containing aluminium silicate (alunite). However, there has been considerable variation in the reporting of lung diseases in relation to exposures to aluminium. Epidemiological studies of workers exposed to aluminium reduction have generally shown low prevalence of pneumoconiotic changes and slight mean reductions in ventilatory lung function. In various work environments, alumina compounds can occur in several forms, and in animal studies these forms appear to have differing lung toxicities. Silica and other mixed dusts may also contribute to this varying toxicity, as may the materials used to coat the aluminium particles. One worker, who developed a granulomatous lung disease after exposure to oxides and metallic aluminium, showed transformation of his blood lymphocytes upon exposure to aluminium salts, suggesting that immunologic factors might play a role.
An asthmatic syndrome has frequently been noted among workers exposed to fumes in aluminium reduction pot rooms. Fluorides found in the pot room environment have been implicated, although the specific agent or agents associated with the asthmatic syndrome has not been determined. As with other occupational asthmas, symptoms are often delayed 4 to 12 hours after exposure, and include cough, dyspnoea, chest tightness and wheeze. An immediate reaction may also be noted. Atopy and a family history of asthma do not appear to be risk factors for development of pot room asthma. After cessation of exposure, symptoms may be expected to disappear in most cases, although two-thirds of the affected workers show persistent non-specific bronchial responsiveness and, in some workers, symptoms and airway hyperresponsiveness continue for years even after exposure is terminated. The prognosis for pot room asthma appears to be best in those who are immediately removed from exposure when the asthmatic symptoms become manifest. Fixed airflow obstruction has also been associated with pot room work.
Carbon electrodes are used in the aluminium reduction process, and known human carcinogens have been identified in the pot room environment. Several mortality studies have revealed lung cancer excesses among exposed workers in this industry.
Diatomaceous Earth
Deposits of diatomaceous earth result from the accretion of skeletons of microscopic organisms. (Cooper and Jacobson 1977; Checkoway et al. 1993.) Diatomaceous earth may be utilized in foundries and in the maintenance of filters, abrasives, lubricants and explosives. Certain deposits comprise up to 90% free silica. Exposed workers may develop lung changes involving simple or complicated pneumoconiosis. The risk of death from both nonmalignant respiratory diseases and lung cancer has been related to the workers’ tenure in dusty work as well as to cumulative crystalline silica exposures during the mining and processing of diatomaceous earth.
Elemental Carbon
Aside from coal, the two common forms of elemental carbon are graphite (crystalline carbon) and carbon black. (Hanoa 1983; Petsonk et al. 1988.) Graphite is used in the manufacture of lead pencils, foundry linings, paints, electrodes, dry batteries and crucibles for metallurgical purposes. Finely ground graphite has lubricant properties. Carbon black is a partially decomposed form used in automotive tires, pigments, plastics, inks and other products. Carbon black is manufactured from fossil fuels through a variety of processes involving partial combustion and thermal decomposition.
Inhalation of carbon, as well as associated dusts, may occur during the mining and milling of natural graphite, and during the manufacture of artificial graphite. Artificial graphite is produced by the heating of coal or petroleum coke, and generally contains no free silica.
Lung health
Pneumoconiosis results from worker exposure to both natural and artificial graphite. Clinically, workers with carbon or graphite pneumoconiosis show radiographic findings similar to those for coal workers. Severe symptomatic cases with massive pulmonary fibrosis were reported in the past, particularly related to the manufacture of carbon electrodes for metallurgy, although recent reports emphasize that the materials implicated in exposures leading to this sort of condition are likely to be mixed dusts.
Gilsonite
Gilsonite, also known as uintaite, is a solidified hydrocarbon. (Keimig et al. 1987.) It occurs in veins in the western United States. Current uses include the manufacture of automotive body seam sealers, inks, paints and enamels. It is an ingredient of oil-well drilling fluids and cements; it is an additive in sand moulds in the foundry industry; it is to be found as a component of asphalt, building boards and explosives; and it is employed in the production of nuclear grade graphite. Workers exposed to gilsonite dust have reported symptoms of cough and phlegm production. Five of ninety-nine workers surveyed showed radiographic evidence of pneumoconiosis. No abnormalities in pulmonary function have been defined in relation to gilsonite dust exposures.
Gypsum
Gypsum is hydrated calcium sulphate (CaSO4·2H2O) (Oakes et al. 1982). It is used as a component of plasterboard, plaster of Paris and Portland cement. Deposits are found in several forms and are often associated with other minerals such as quartz. Pneumoconiosis has been observed in gypsum miners, and has been attributed to silica contamination. Ventilatory abnormalities have not been associated with gypsum dust exposures.
Oils and Lubricants
Liquids containing hydrocarbon oils are used as coolants, cutting oils and lubricants (Cullen et al. 1981). Vegetable oils are found in some commercial products and in a variety of foodstuffs. These oils may be aerosolized and inhaled when metals that are coated with oils are milled or machined, or if oil-containing sprays are used for purposes of cleaning or lubrication. Environmental measurements in machine shops and mills have documented airborne oil levels up to 9 mg/m3. One report implicated airborne oil exposure from the burning of animal and vegetable fats in an enclosed building.
Lung health
Workers exposed to these aerosols have occasionally been reported to develop evidence of a lipoid pneumonia, similar to that noted in patients who have aspirated mineral oil nose drops or other oily materials. The condition is associated with symptoms of cough and dyspnoea, inspiratory lung crackles, and impairments in lung function, generally mild in severity. A few cases have been reported with more extensive radiographic changes and severe lung impairments. Exposure to mineral oils has also been associated in several studies with an increased risk of respiratory tract cancers.
Portland Cement
Portland cement is made from hydrated calcium silicates, aluminium oxide, magnesium oxide, iron oxide, calcium sulphate, clay, shale and sand (Abrons et al. 1988; Yan et al. 1993). The mixture is crushed and calcined at high temperatures with the addition of gypsum. Cement finds numerous uses in road and building construction.
Lung health
Silicosis appears to be the greatest risk in cement workers, followed by a mixed dust pneumoconiosis. (In the past, asbestos was added to cement to improve its characteristics.) Abnormal chest radiographic findings, including small rounded and irregular opacities and pleural changes, have been noted. Workers have occasionally been reported to have developed pulmonary alveolar proteinosis after the inhalation of cement dust. Airflow obstructive changes have been noted in some, but not all, surveys of cement workers.
Rare Earth Metals
Rare earth metals or “lanthanides” have atomic numbers between 57 and 71. Lanthanum (atomic number 57), cerium (58), and neodymium (60) are the commonest of the group. The other elements in this group include praseodymium (59), promethium (61), samarium (62), europium (63), gadolinium (64), terbium (65), dysprosium (66), holmium (67), erbium (68), thulium (69), ytterbium (70) and lutetium (71). (Hussain, Dick and Kaplan 1980; Sabbioni, Pietra and Gaglione 1982; Vocaturo, Colombo and Zanoni 1983; Sulotto, Romano and Berra 1986; Waring and Watling 1990; Deng et al. 1991.) The rare earth elements are found naturally in monazite sand, from which they are extracted. They are used in a variety of alloy metals, as abrasives for polishing mirrors and lenses, for high-temperature ceramics, in fireworks and in cigarette lighter flints. In the electronics industry they are used in electrowelding and are to be found in various electronic components, including television phosphors, radiographic screens, lasers, microwave devices, insulators, capacitors and semiconductors.
Carbon arc lamps are used widely in the printing, photoengraving and lithography industries and were used for floodlighting, spotlighting and movie projection before the wide-scale adoption of argon and xenon lamps. The rare earth metal oxides were incorporated into the central core of carbon arc rods, where they stabilize the arc stream. Fumes which are emitted from the lamps are a mixture of gaseous and particulate material composed of approximately 65% rare earth oxides, 10% fluorides and unburnt carbon and impurities.
Lung health
Pneumoconiosis in workers exposed to rare earths has been exhibited primarily as bilateral nodular chest radiographic infiltrates. Lung pathology in cases of rare earth pneumoconiosis has been described as an interstitial fibrosis accompanied by an accumulation of fine granular dust particles, or granulomatous changes.
Variable pulmonary function impairments have been described, from restrictive to mixed restrictive-obstructive. However, the spectrum of pulmonary disease related to inhalation of rare earth elements is still to be defined, and data regarding the pattern and progression of disease and histological changes is available primarily only from a few case reports.
A neoplastic potential of the rare earth isotopes has been suggested by a case report of lung cancer, possibly related to ionizing radiation from the naturally occurring rare earth radioisotopes.
Sedimentary Compounds
Sedimentary rock deposits form through the processes of physical and chemical weathering, erosion, transport, deposition and diagenesis. These may be characterized into two broad classes: Clastics, which include mechanically deposited erosion debris, and chemical precipitates, which include carbonates, shells of organic skeletons and saline deposits. Sedimentary carbonates, sulphates and halides provide relatively pure minerals that have crystallized from concentrated solutions. Due to the high solubility of many of the sedimentary compounds, they are rapidly cleared from the lungs and are generally associated with little pulmonary pathology. In contrast, workers exposed to certain sedimentary compounds, primarily clastics, have shown pneumoconiotic changes.
Phosphates
Phosphate ore, Ca5(F,Cl)(PO4)3, is used in the production of fertilizers, dietary supplements, toothpaste, preservatives, detergents, pesticides, rodent poisons and ammunitions (Dutton et al. 1993). Extraction and processing of the ore may result in a variety of irritant exposures. Surveys of workers in phosphate mining and extraction have documented increased symptoms of cough and phlegm production, as well as radiographic evidence of pneumoconiosis, but little evidence of abnormal lung function.
Shale
Shale is a mixture of organic material composed mainly of carbon, hydrogen, oxygen, sulphur and nitrogen (Rom, Lee and Craft 1981; Seaton et al. 1981). The mineral component (kerogen) is found in the sedimentary rock called marlstone, which is of a grey-brown colour and a layered consistency. Oil shale has been used as an energy source since the 1850s in Scotland. Major deposits exist in the United States, Scotland and Estonia. Dust in the atmosphere of underground oil shale mines is of relatively fine dispersion, with up to 80% of the dust particles under 2 mm in size.
Lung health
Pneumoconiosis related to the deposition of shale dust in the lung is termed shalosis. The dust creates a granulomatous and fibrotic reaction in the lungs. This pneumoconiosis is similar clinically to coal workers’ pneumoconiosis and silicosis, and may progress to massive fibrosis even after the worker has left the industry.
Pathologic changes identified in lungs with shalosis are characterized by vascular and bronchial deformation, with irregular thickening of interalveolar and interlobular septa. In addition to interstitial fibrosis, lung specimens with shale pneumoconiosis have shown enlarged hilar shadows, related to the transport of shale dust and subsequent development of well-defined sclerotic changes in the hilar lymph nodes.
Shale workers have been found to have a prevalence of chronic bronchitis two and one-half times that of age-matched controls. The effect of shale dust exposures on lung function has not been studied systematically.
Slate
Slate is a metamorphic rock, made up of various minerals, clays and carbonaceous matter (McDermott et al. 1978). The major constituents of slate include muscovite, chlorite, calcite and quartz, along with graphite, magnetite and rutile. These have undergone metamorphosis to form a dense crystalline rock that possesses strength but is easily cleaved, characteristics which account for its economic importance. Slate is used in roofing, dimension stone, floor tile, flagging, structural shapes such as panels and window sills, blackboards, pencils, billiard tables and laboratory bench tops. Crushed slate is used in highway construction, tennis court surfaces and lightweight roofing granules.
Lung health
Pneumoconiosis has been found in a third of workers studied in the slate industry in North Wales, and in 54% of slate pencil makers in India. Various lung radiographic changes have been identified in slateworkers. Because of the high quartz content of some slates and the adjacent rock strata, slateworkers’ pneumoconiosis may have features of silicosis. The prevalence of respiratory symptoms in slateworkers is high, and the proportion of workers with symptoms increases with pneumoconiosis category, irrespective of smoking status. Diminished values of forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) are associated with increasing pneumoconiosis category.
The lungs of miners exposed to slate dust reveal localized areas of perivascular and peribronchial fibrosis, extending to macule formation and extensive interstitial fibrosis. Typical lesions are fibrotic macules of variable configuration intimately associated with small pulmonary blood vessels.
Talc
Talc is composed of magnesium silicates, and is found in a variety of forms. (Vallyathan and Craighead 1981; Wegman et al. 1982; Stille and Tabershaw 1982; Wergeland, Andersen and Baerheim 1990; Gibbs, Pooley and Griffith 1992.)
Deposits of talc are frequently contaminated with other minerals, including both fibrous and non-fibrous tremolite and quartz. Lung health effects of talc-exposed workers may be related to both the talc itself as well as the other associated minerals.
Talc production occurs primarily in Australia, Austria, China, France and the United States. Talc is used as a component in hundreds of products, and is used in the manufacture of paint, pharmaceuticals, cosmetics, ceramics, automobile tires and paper.
Lung health
Diffuse rounded and irregular parenchymal lung opacities and pleural abnormalities are seen on the chest radiographs of talc workers in association with the talc exposure. Depending on the specific exposures experienced, the radiographic shadows may be ascribed to talc itself or to contaminants in the talc. Talc exposure has been associated with symptoms of cough, dyspnoea and phlegm production, and with evidence of airflow obstruction in pulmonary function studies. Lung pathology has revealed various forms of pulmonary fibrosis: granulomatous changes and ferruginous bodies have been reported, and dust-laden macrophages collected around the respiratory bronchioles intermingled with bundles of collagen. Mineralogical examination of lung tissue from talc workers is also variable and may show silica, mica or mixed silicates.
Since talc deposits may be associated with asbestos and other fibres, it is not surprising that an increased risk of bronchogenic carcinoma has been reported in talc miners and millers. Recent investigations of workers exposed to talc without associated asbestos fibres revealed trends for higher mortality from non-malignant respiratory disease (silicosis, silico-tuberculosis, emphysema and pneumonia), but the risk for bronchogenic cancer was not found to be elevated.
Hairspray
Exposure to hairsprays occurs in the home environment as well as in commercial hairdressing establishments (Rom 1992b). Environmental measurements in beauty salons have indicated the potential for respirable aerosol exposures. Several case reports have implicated hairspray exposure in the occurrence of a pneumonitis, thesaurosis, in heavily exposed individuals. Clinical symptoms in the cases were generally mild, and resolved with termination of exposure. Histology usually showed a granulomatous process in the lung and enlarged hilar lymph nodes, with thickening of alveolar walls and numerous granular macrophages in the airspaces. Macromolecules in hairsprays, including shellacs and polyvinylpyrrolidone, have been suggested as potential agents. In contrast to the clinical case reports, increased lung parenchymal radiographic shadows observed in radiological surveys of commercial hairdressers have not been conclusively related to hairspray exposure. Although the results of these studies do not allow definitive conclusions to be drawn, clinically important lung disease from typical hairspray exposures does appear to be an unusual occurrence.
" DISCLAIMER: The ILO does not take responsibility for content presented on this web portal that is presented in any language other than English, which is the language used for the initial production and peer-review of original content. Certain statistics have not been updated since the production of the 4th edition of the Encyclopaedia (1998)."