It has long been recognized that each person’s response to environmental chemicals is different. The recent explosion in molecular biology and genetics has brought a clearer understanding about the molecular basis of such variability. Major determinants of individual response to chemicals include important differences among more than a dozen superfamilies of enzymes, collectively termed xenobiotic- (foreign to the body) or drug-metabolizing enzymes. Although the role of these enzymes has classically been regarded as detoxification, these same enzymes also convert a number of inert compounds to highly toxic intermediates. Recently, many subtle as well as gross differences in the genes encoding these enzymes have been identified, which have been shown to result in marked variations in enzyme activity. It is now clear that each individual possesses a distinct complement of xenobiotic-metabolizing enzyme activities; this diversity might be thought of as a “metabolic fingerprint”. It is the complex interplay of these many different enzyme superfamilies which ultimately determines not only the fate and the potential for toxicity of a chemical in any given individual, but also assessment of exposure. In this article we have chosen to use the cytochrome P450 enzyme superfamily to illustrate the remarkable progress made in understanding individual response to chemicals. The development of relatively simple DNA-based tests designed to identify specific gene alterations in these enzymes, is now providing more accurate predictions of individual response to chemical exposure. We hope the result will be preventive toxicology. In other words, each individual might learn about those chemicals to which he or she is particularly sensitive, thereby avoiding previously unpredictable toxicity or cancer.
Although it is not generally appreciated, human beings are exposed daily to a barrage of innumerable diverse chemicals. Many of these chemicals are highly toxic, and they are derived from a wide variety of environmental and dietary sources. The relationship between such exposures and human health has been, and continues to be, a major focus of biomedical research efforts worldwide.
What are some examples of this chemical bombardment? More than 400 chemicals from red wine have been isolated and characterized. At least 1,000 chemicals are estimated to be produced by a lighted cigarette. There are countless chemicals in cosmetics and perfumed soaps. Another major source of chemical exposure is agriculture: in the United States alone, farmlands receive more than 75,000 chemicals each year in the form of pesticides, herbicides and fertilizing agents; after uptake by plants and grazing animals, as well as fish in nearby waterways, humans (at the end of the food chain) ingest these chemicals. Two other sources of large concentrations of chemicals taken into the body include (a) drugs taken chronically and (b) exposure to hazardous substances in the workplace over a lifetime of employment.
It is now well established that chemical exposure may adversely affect many aspects of human health, causing chronic diseases and the development of many cancers. In the last decade or so, the molecular basis of many of these relationships has begun to be unravelled. In addition, the realization has emerged that humans differ markedly in their susceptibility to the harmful effects of chemical exposure.
Current efforts to predict human response to chemical exposure combine two fundamental approaches (figure 1): monitoring the extent of human exposure through biological markers (biomarkers), and predicting the likely response of an individual to a given level of exposure. Although both of these approaches are extremely important, it should be emphasized that the two are distinctly different from one another. This article will focus on the genetic factors underlying individual susceptibility to any particular chemical exposure. This field of research is broadly termed ecogenetics, or pharmacogenetics (see Kalow 1962 and 1992). Many of the recent advances in determining individual susceptibility to chemical toxicity have evolved from a greater appreciation of the processes by which humans and other mammals detoxify chemicals, and the remarkable complexity of the enzyme systems involved.
Figure 1. The interrelationships among exposure assessment, ethnic differences, age, diet, nutrition and genetic susceptibility assessment - all of which play a role in the individual risk of toxicity and cancer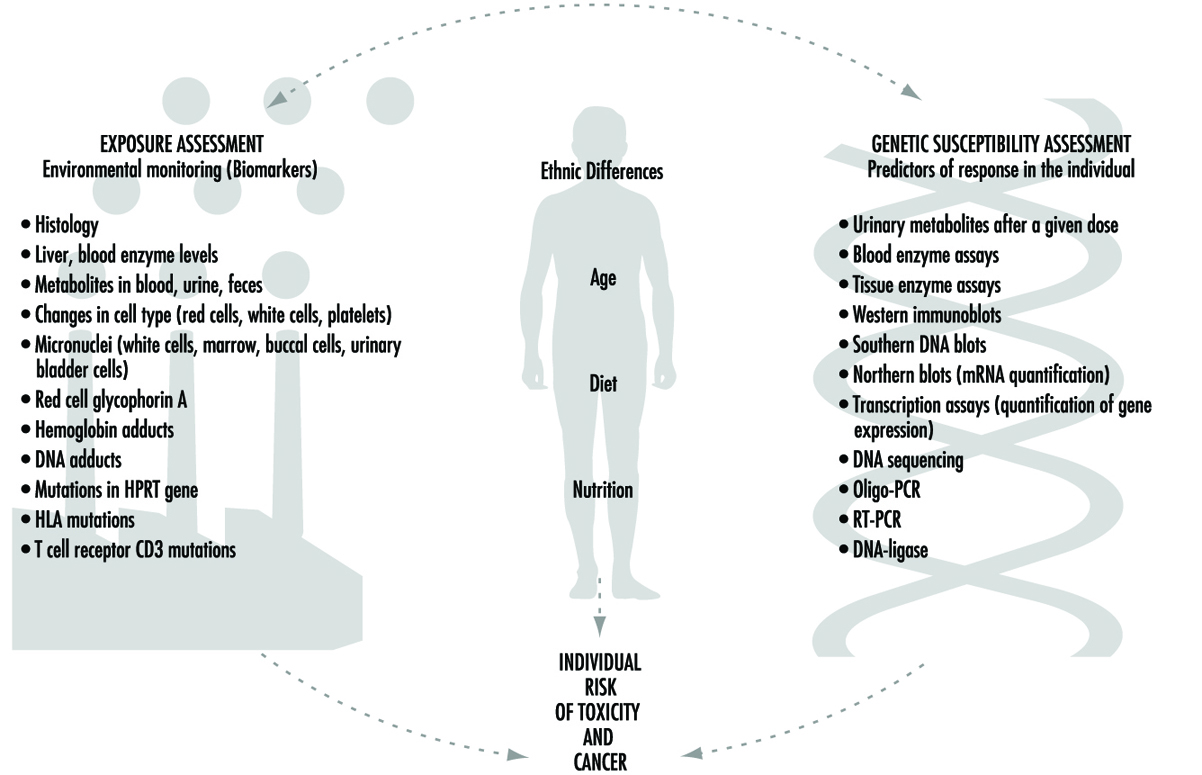
We will first describe the variability of toxic responses in humans. We will then introduce some of the enzymes responsible for such variation in response, due to differences in the metabolism of foreign chemicals. Next, the history and nomenclature of the cytochrome P450 superfamily will be detailed. Five human P450 polymorphisms as well as several non-P450 polymorphisms will be briefly described; these are responsible for human differences in toxic response. We will then discuss an example to emphasize the point that genetic differences in individuals can influence exposure assessment, as determined by environmental monitoring. Lastly, we will discuss the role of these xenobiotic-metabolizing enzymes in critical life functions.
Variation in Toxic Response Among the Human Population
Toxicologists and pharmacologists commonly speak about the average lethal dose for 50% of the population (LD50), the average maximal tolerated dose for 50% of the population (MTD50), and the average effective dose of a particular drug for 50% of the population (ED50). However, how do these doses affect each of us on an individual basis? In other words, a highly sensitive individual may be 500 times more affected or 500 times more likely to be affected than the most resistant individual in a population; for these people, the LD50 (and MTD50 and ED50) values would have little meaning. LD50, MTD50 and ED50 values are only relevant when referring to the population as a whole.
Figure 2 illustrates a hypothetical dose-response relationship for a toxic response by individuals in any given population. This generic diagram might represent bronchogenic carcinoma in response to the number of cigarettes smoked, chloracne as a function of dioxin levels in the workplace, asthma as a function of air concentrations of ozone or aldehyde, sunburn in response to ultraviolet light, decreased clotting time as a function of aspirin intake, or gastrointestinal distress in response to the number of jalapeño peppers consumed. Generally, in each of these instances, the greater the exposure, the greater the toxic response. Most of the population will exhibit the mean and standard deviation of toxic response as a function of dose. The “resistant outlier” (lower right in figure 2) is an individual having less of a response at higher doses or exposures. A “sensitive outlier” (upper left) is an individual having an exaggerated response to a relatively small dose or exposure. These outliers, with extreme differences in response compared to the majority of individuals in the population, may represent important genetic variants that can help scientists in attempting to understand the underlying molecular mechanisms of a toxic response.
Figure 2. Generic relationship between any toxic response and the dose of any environmental, chemical or physical agent
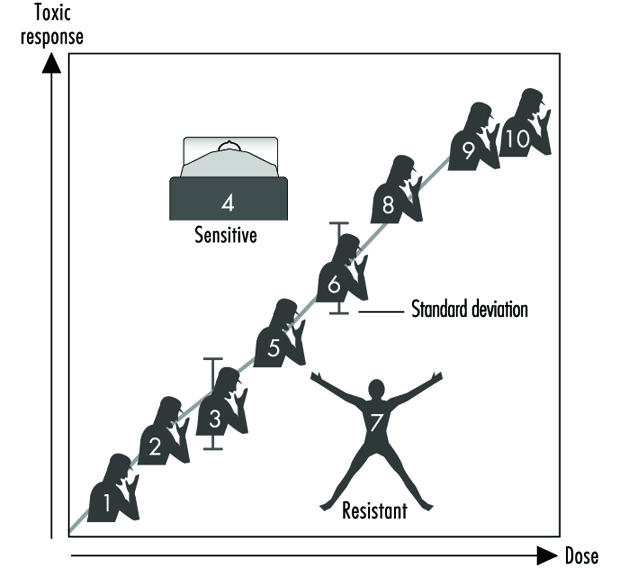
Using these outliers in family studies, scientists in a number of laboratories have begun to appreciate the importance of Mendelian inheritance for a given toxic response. Subsequently, one can then turn to molecular biology and genetic studies to pinpoint the underlying mechanism at the gene level (genotype) responsible for the environmentally caused disease (phenotype).
Xenobiotic- or Drug-metabolizing Enzymes
How does the body respond to the myriad of exogenous chemicals to which we are exposed? Humans and other mammals have evolved highly complex metabolic enzyme systems comprising more than a dozen distinct superfamilies of enzymes. Almost every chemical to which humans are exposed will be modified by these enzymes, in order to facilitate removal of the foreign substance from the body. Collectively, these enzymes are frequently referred to as drug-metabolizing enzymes or xenobiotic-metabolizing enzymes. Actually, both terms are misnomers. First, many of these enzymes not only metabolize drugs but hundreds of thousands of environmental and dietary chemicals. Second, all of these enzymes also have normal body compounds as substrates; none of these enzymes metabolizes only foreign chemicals.
For more than four decades, the metabolic processes mediated by these enzymes have commonly been classified as either Phase I or Phase II reactions (figure 3). Phase I (“functionalization”) reactions generally involve relatively minor structural modifications of the parent chemical via oxidation, reduction or hydrolysis in order to produce a more water-soluble metabolite. Frequently, Phase I reactions provide a “handle” for further modification of a compound by subsequent Phase II reactions. Phase I reactions are primarily mediated by a superfamily of highly versatile enzymes, collectively termed cytochromes P450, although other enzyme superfamilies can also be involved (figure 4).
Figure 3. The classical designation of Phase I and Phase II xenobiotic- or drug-metabolizing enzymes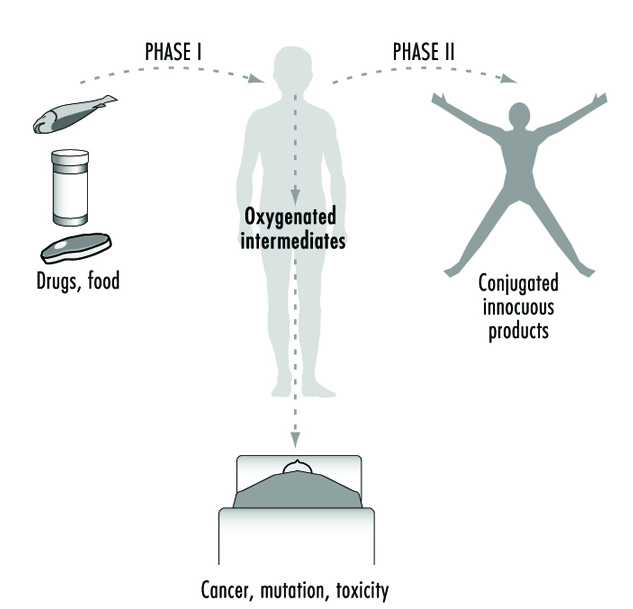
Figure 4. Examples of drug-metabolizing enzymes
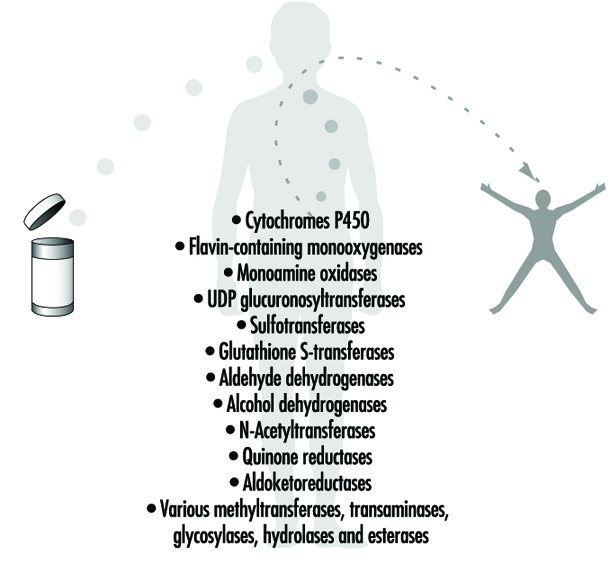
Phase II reactions involve the coupling of a water-soluble endogenous molecule to a chemical (parent chemical or Phase I metabolite) in order to facilitate excretion. Phase II reactions are frequently termed “conjugation” or “derivatization” reactions. The enzyme superfamilies catalyzing Phase II reactions are generally named according to the endogenous conjugating moiety involved: for example, acetylation by the N-acetyltransferases, sulphation by the sulphotransferases, glutathione conjugation by the glutathione transferases, and glucuronidation by the UDP glucuronosyltransferases (figure 4). Although the major organ of drug metabolism is the liver, the levels of some drug- metabolizing enzymes are quite high in the gastrointestinal tract, gonads, lung, brain and kidney, and such enzymes are undoubtedly present to some extent in every living cell.
Xenobiotic-metabolizing Enzymes Represent Double-edged Swords
As we learn more about the biological and chemical processes leading to human health aberrations, it has become increasingly evident that drug-metabolizing enzymes function in an ambivalent manner (figure 3). In the majority of cases, lipid-soluble chemicals are converted to more readily excreted water-soluble metabolites. However, it is clear that on many occasions the same enzymes are capable of transforming other inert chemicals into highly reactive molecules. These intermediates can then interact with cellular macromolecules such as proteins and DNA. Thus, for each chemical to which humans are exposed, there exists the potential for the competing pathways of metabolic activation and detoxification.
Brief Review of Genetics
In human genetics, each gene (locus) is located on one of the 23 pairs of chromosomes. The two alleles (one present on each chromosome of the pair) can be the same, or they can be different from one another. For example, the B and b alleles, in which B (brown eyes) is dominant over b (blue eyes): individuals of the brown-eyed phenotype can have either the BB or Bb genotypes, whereas individuals of the blue-eyed phenotype can only have the bb genotype.
A polymorphism is defined as two or more stably inherited phenotypes (traits)—derived from the same gene(s)—that are maintained in the population, often for reasons not necessarily obvious. For a gene to be polymorphic, the gene product must not be essential for development, reproductive vigour or other critical life processes. In fact, a “balanced polymorphism,” wherein the heterozygote has a distinct survival advantage over either homozygote (e.g., resistance to malaria, and the sickle-cell haemoglobin allele) is a common explanation for maintaining an allele in the population at otherwise unexplained high frequencies (see Gonzalez and Nebert 1990).
Human Polymorphisms of Xenobiotic-metabolizing Enzymes
Genetic differences in the metabolism of various drugs and environmental chemicals have been known for more than four decades (Kalow 1962 and 1992). These differences are frequently referred to as pharmacogenetic or, more broadly, ecogenetic polymorphisms. These polymorphisms represent variant alleles that occur at a relatively high frequency in the population and are generally associated with aberrations in enzyme expression or function. Historically, polymorphisms were usually identified following unexpected responses to therapeutic agents. More recently, recombinant DNA technology has enabled scientists to identify the precise alterations in genes that are responsible for some of these polymorphisms. Polymorphisms have now been characterized in many drug-metabolizing enzymes—including both Phase I and Phase II enzymes. As more and more polymorphisms are identified, it is becoming increasingly apparent that each individual may possess a distinct complement of drug-metabolizing enzymes. This diversity might be described as a “metabolic fingerprint”. It is the complex interplay of the various drug- metabolizing enzyme superfamilies within any individual that will ultimately determine his or her particular response to a given chemical (Kalow 1962 and 1992; Nebert 1988; Gonzalez and Nebert 1990; Nebert and Weber 1990).
Expressing Human Xenobiotic-metabolizingEnzymes in Cell Culture
How might we develop better predictors of human toxic responses to chemicals? Advances in defining the multiplicity of drug-metabolizing enzymes must be accompanied by precise knowledge as to which enzymes determine the metabolic fate of individual chemicals. Data gleaned from laboratory rodent studies have certainly provided useful information. However, significant interspecies differences in xenobiotic-metabolizing enzymes necessitate caution in extrapolating data to human populations. To overcome this difficulty, many laboratories have developed systems in which various cell lines in culture can be engineered to produce functional human enzymes that are stable and in high concentrations (Gonzalez, Crespi and Gelboin 1991). Successful production of human enzymes has been achieved in a variety of diverse cell lines from sources including bacteria, yeast, insects and mammals.
In order to define the metabolism of chemicals even more accurately, multiple enzymes have also been successfully produced in a single cell line (Gonzalez, Crespi and Gelboin 1991). Such cell lines provide valuable insights into the precise enzymes involved in the metabolic processing of any given compound and likely toxic metabolites. If this information can then be combined with knowledge regarding the presence and level of an enzyme in human tissues, these data should provide valuable predictors of response.
Cytochrome P450
History and nomenclature
The cytochrome P450 superfamily is one of the most studied drug-metabolizing enzyme superfamilies, having a great deal of individual variability in response to chemicals. Cytochrome P450 is a convenient generic term used to describe a large superfamily of enzymes pivotal in the metabolism of innumerable endogenous and exogenous substrates. The term cytochrome P450 was first coined in 1962 to describe an unknown pigment in cells which, when reduced and bound with carbon monoxide, produced a characteristic absorption peak at 450 nm. Since the early 1980s, cDNA cloning technology has resulted in remarkable insights into the multiplicity of cytochrome P450 enzymes. To date, more than 400 distinct cytochrome P450 genes have been identified in animals, plants, bacteria and yeast. It has been estimated that any one mammalian species, such as humans, may possess 60 or more distinct P450 genes (Nebert and Nelson 1991). The multiplicity of P450 genes has necessitated the development of a standardized nomenclature system (Nebert et al. 1987; Nelson et al. 1993). First proposed in 1987 and updated on a biannual basis, the nomenclature system is based on divergent evolution of amino acid sequence comparisons between P450 proteins. The P450 genes are divided into families and subfamilies: enzymes within a family display greater than 40% amino acid similarity, and those within the same subfamily display 55% similarity. P450 genes are named with the root symbol CYP followed by an arabic numeral designating the P450 family, a letter denoting the subfamily, and a further arabic numeral designating the individual gene (Nelson et al. 1993; Nebert et al. 1991). Thus, CYP1A1 represents P450 gene 1 in family 1 and subfamily A.
As of February 1995, there are 403 CYP genes in the database, composed of 59 families and 105 sub- families. These include eight lower eukaryotic families, 15 plant families, and 19 bacterial families. The 15 human P450 gene families comprise 26 subfamilies, 22 of which have been mapped to chromosomal locations throughout most of the genome. Some sequences are clearly orthologous across many species—for example, only one CYP17 (steroid 17α-hydroxylase) gene has been found in all vertebrates examined to date; other sequences within a subfamily are highly duplicated, making the identification of orthologous pairs impossible (e.g., the CYP2C subfamily). Interestingly, human and yeast share an orthologous gene in the CYP51 family. Numerous comprehensive reviews are available for readers seeking further information on the P450 superfamily (Nelson et al. 1993; Nebert et al. 1991; Nebert and McKinnon 1994; Guengerich 1993; Gonzalez 1992).
The success of the P450 nomenclature system has resulted in similar terminology systems being developed for the UDP glucuronosyltransferases (Burchell et al. 1991) and flavin-containing mono-oxygenases (Lawton et al. 1994). Similar nomenclature systems based on divergent evolution are also under development for several other drug-metabolizing enzyme superfamilies (e.g., sulphotransferases, epoxide hydrolases and aldehyde dehydrogenases).
Recently, we divided the mammalian P450 gene superfamily into three groups (Nebert and McKinnon 1994)—those involved principally with foreign chemical metabolism, those involved in the synthesis of various steroid hormones, and those participating in other important endogenous functions. It is the xenobiotic-metabolizing P450 enzymes that assume the most significance for prediction of toxicity.
Xenobiotic-metabolizing P450 enzymes
P450 enzymes involved in the metabolism of foreign compounds and drugs are almost always found within families CYP1, CYP2, CYP3 and CYP4. These P450 enzymes catalyze a wide variety of metabolic reactions, with a single P450 often capable of meta-bolizing many different compounds. In addition, multiple P450 enzymes may metabolize a single compound at different sites. Also, a compound may be metabolized at the same, single site by several P450s, although at varying rates.
A most important property of the drug-metabolizing P450 enzymes is that many of these genes are inducible by the very substances which serve as their substrates. On the other hand, other P450 genes are induced by nonsubstrates. This phenomenon of enzyme induction underlies many drug-drug interactions of therapeutic importance.
Although present in many tissues, these particular P450 enzymes are found in relatively high levels in the liver, the primary site of drug metabolism. Some of the xenobiotic-metabolizing P450 enzymes exhibit activity toward certain endogenous substrates (e.g., arachidonic acid). However, it is generally believed that most of these xenobiotic-metabolizing P450 enzymes do not play important physiological roles—although this has not been established experimentally as yet. The selective homozygous disruption, or “knock-out,” of individual xenobiotic-metabolizing P450 genes by means of gene targeting methodologies in mice is likely to provide unequivocal information soon with regard to physiological roles of the xenobiotic-metabolizing P450s (for a review of gene targeting, see Capecchi 1994).
In contrast to P450 families encoding enzymes involved primarily in physiological processes, families encoding xenobiotic-metabolizing P450 enzymes display marked species specificity and frequently contain many active genes per subfamily (Nelson et al. 1993; Nebert et al. 1991). Given the apparent lack of physiological substrates, it is possible that P450 enzymes in families CYP1, CYP2, CYP3 and CYP4 that have appeared in the past several hundred million years have evolved as a means of detoxifying foreign chemicals encountered in the environment and diet. Clearly, evolution of the xenobiotic-metabolizing P450s would have occurred over a time period which far precedes the synthesis of most of the synthetic chemicals to which humans are now exposed. The genes in these four gene families may have evolved and diverged in animals due to their exposure to plant metabolites during the last 1.2 billion years—a process descriptively termed “animal-plant warfare” (Gonzalez and Nebert 1990). Animal-plant warfare is the phenomenon in which plants developed new chemicals (phytoalexins) as a defence mechanism in order to prevent ingestion by animals, and animals, in turn, responded by developing new P450 genes to accommodate the diversifying substrates. Providing further impetus to this proposal are the recently described examples of plant-insect and plant-fungus chemical warfare involving P450 detoxification of toxic substrates (Nebert 1994).
The following is a brief introduction to several of the human xenobiotic-metabolizing P450 enzyme polymorphisms in which genetic determinants of toxic response are believed to be of high significance. Until recently, P450 polymorphisms were generally suggested by unexpected variance in patient response to administered therapeutic agents. Several P450 polymorphisms are indeed named according to the drug with which the polymorphism was first identified. More recently, research efforts have focused on identification of the precise P450 enzymes involved in the metabolism of chemicals for which variance is observed and the precise characterization of the P450 genes involved. As described earlier, the measurable activity of a P450 enzyme towards a model chemical can be called the phenotype. Allelic differences in a P450 gene for each individual is termed the P450 genotype. As more and more scrutiny is applied to the analysis of P450 genes, the precise molecular basis of previously documented phenotypic variance is becoming clearer.
The CYP1A subfamily
The CYP1A subfamily comprises two enzymes in humans and all other mammals: these are designated CYP1A1 and CYP1A2 under standard P450 nomenclature. These enzymes are of considerable interest, because they are involved in the metabolic activation of many procarcinogens and are also induced by several compounds of toxicological concern, including dioxin. For example, CYP1A1 metabolically activates many compounds found in cigarette smoke. CYP1A2 metabolically activates many arylamines—associated with urinary bladder cancer—found in the chemical dye industry. CYP1A2 also metabolically activates 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a tobacco-derived nitrosamine. CYP1A1 and CYP1A2 are also found at higher levels in the lungs of cigarette smokers, due to induction by polycyclic hydrocarbons present in the smoke. The levels of CYP1A1 and CYP1A2 activity are therefore considered to be important determinants of individual response to many potentially toxic chemicals.
Toxicological interest in the CYP1A subfamily was greatly intensified by a 1973 report correlating the level of CYP1A1 inducibility in cigarette smokers with individual susceptibility to lung cancer (Kellermann, Shaw and Luyten-Kellermann 1973). The molecular basis of CYP1A1 and CYP1A2 induction has been a major focus of numerous laboratories. The induction process is mediated by a protein termed the Ah receptor to which dioxins and structurally related chemicals bind. The name Ah is derived from the aryl hydrocarbon nature of many CYP1A inducers. Interestingly, differences in the gene encoding the Ah receptor between strains of mice result in marked differences in chemical response and toxicity. A polymorphism in the Ah receptor gene also appears to occur in humans: approximately one-tenth of the population displays high induction of CYP1A1 and may be at greater risk than the other nine-tenths of the population for development of certain chemically induced cancers. The role of the Ah receptor in the control of enzymes in the CYP1A subfamily, and its role as a determinant of human response to chemical exposure, has been the subject of several recent reviews (Nebert, Petersen and Puga 1991; Nebert, Puga and Vasiliou 1993).
Are there other polymorphisms that might control the level of CYP1A proteins in a cell? A polymorphism in the CYP1A1 gene has also been identified, and this appears to influence lung cancer risk amongst Japanese cigarette smokers, although this same polymorphism does not appear to influence risk in other ethnic groups (Nebert and McKinnon 1994).
CYP2C19
Variations in the rate at which individuals metabolize the anticonvulsant drug (S)-mephenytoin have been well documented for many years (Guengerich 1989). Between 2% and 5% of Caucasians and as many as 25% of Asians are deficient in this activity and may be at greater risk of toxicity from the drug. This enzyme defect has long been known to involve a member of the human CYP2C subfamily, but the precise molecular basis of this deficiency has been the subject of considerable controversy. The major reason for this difficulty was the six or more genes in the human CYP2C subfamily. It was recently demonstrated, however, that a single-base mutation in the CYP2C19 gene is the primary cause of this deficiency (Goldstein and de Morais 1994). A simple DNA test, based on the polymerase chain reaction (PCR), has also been developed to identify this mutation rapidly in human populations (Goldstein and de Morais 1994).
CYP2D6
Perhaps the most extensively characterized variation in a P450 gene is that involving the CYP2D6 gene. More than a dozen examples of mutations, rearrangements and deletions affecting this gene have been described (Meyer 1994). This polymorphism was first suggested 20 years ago by clinical variability in patients’ response to the antihypertensive agent debrisoquine. Alterations in the CYP2D6 gene giving rise to altered enzyme activity are therefore collectively termed the debrisoquine polymorphism.
Prior to the advent of DNA-based studies, individuals had been classified as poor or extensive metabolizers (PMs, EMs) of debrisoquine based on metabolite concentrations in urine samples. It is now clear that alterations in the CYP2D6 gene may result in individuals displaying not only poor or extensive debrisoquine metabolism, but also ultrarapid metabolism. Most alterations in the CYP2D6 gene are associated with partial or total deficiency of enzyme function; however, individuals in two families have recently been described who possess multiple functional copies of the CYP2D6 gene, giving rise to ultrarapid metabolism of CYP2D6 substrates (Meyer 1994). This remarkable observation provides new insights into the wide spectrum of CYP2D6 activity previously observed in population studies. Alterations in CYP2D6 function are of particular significance, given the more than 30 commonly prescribed drugs metabolized by this enzyme. An individual’s CYP2D6 function is therefore a major determinant of both therapeutic and toxic response to administered therapy. Indeed, it has recently been argued that consideration of a patient’s CYP2D6 status is necessary for the safe use of both psychiatric and cardiovascular drugs.
The role of the CYP2D6 polymorphism as a determinant of individual susceptibility to human diseases such as lung cancer and Parkinson’s disease has also been the subject of intense study (Nebert and McKinnon 1994; Meyer 1994). While conclusions are difficult to define given the diverse nature of the study protocols utilized, the majority of studies appear to indicate an association between extensive metabolizers of debrisoquine (EM phenotype) and lung cancer. The reasons for such an association are presently unclear. However, the CYP2D6 enzyme has been shown to metabolize NNK, a tobacco-derived nitrosamine.
As DNA-based assays improve—enabling even more accurate assessment of CYP2D6 status—it is anticipated that the precise relationship of CYP2D6 to disease risk will be clarified. Whereas the extensive metabolizer may be linked with susceptibility to lung cancer, the poor metabolizer (PM phenotype) appears to be associated with Parkinson’s disease of unknown cause. Whereas these studies are also difficult to compare, it appears that PM individuals having a diminished capacity to metabolize CYP2D6 substrates (e.g., debrisoquine) have a 2- to 2.5-fold increase in risk of developing Parkinson’s disease.
CYP2E1
The CYP2E1 gene encodes an enzyme that metabolizes many chemicals, including drugs and many low-molecular-weight carcinogens. This enzyme is also of interest because it is highly inducible by alcohol and may play a role in liver injury induced by chemicals such as chloroform, vinyl chloride and carbon tetrachloride. The enzyme is primarily found in the liver, and the level of enzyme varies markedly between individuals. Close scrutiny of the CYP2E1 gene has resulted in the identification of several polymorphisms (Nebert and McKinnon 1994). A relationship has been reported between the presence of certain structural variations in the CYP2E1 gene and apparent lowered lung cancer risk in some studies; however, there are clear interethnic differences which require clarification of this possible relationship.
The CYP3A subfamily
In humans, four enzymes have been identified as members of the CYP3A subfamily due to their similarity in amino acid sequence. The CYP3A enzymes metabolize many commonly prescribed drugs such as erythromycin and cyclosporin. The carcinogenic food contaminant aflatoxin B1 is also a CYP3A substrate. One member of the human CYP3A subfamily, designated CYP3A4, is the principal P450 in human liver as well as being present in the gastrointestinal tract. As is true for many other P450 enzymes, the level of CYP3A4 is highly variable between individuals. A second enzyme, designated CYP3A5, is found in only approximately 25% of livers; the genetic basis of this finding has not been elucidated. The importance of CYP3A4 or CYP3A5 variability as a factor in genetic determinants of toxic response has not yet been established (Nebert and McKinnon 1994).
Non-P450 Polymorphisms
Numerous polymorphisms also exist within other xenobiotic-metabolizing enzyme superfamilies (e.g., glutathione transferases, UDP glucuronosyltransferases, para-oxonases, dehydrogenases, N-acetyltransferases and flavin-containing mono-oxygenases). Because the ultimate toxicity of any P450-generated intermediate is dependent on the efficiency of subsequent Phase II detoxification reactions, the combined role of multiple enzyme polymorphisms is important in determining susceptibility to chemically induced diseases. The metabolic balance between Phase I and Phase II reactions (figure 3) is therefore likely to be a major factor in chemically induced human diseases and genetic determinants of toxic response.
The GSTM1 gene polymorphism
A well studied example of a polymorphism in a Phase II enzyme is that involving a member of the glutathione S-transferase enzyme superfamily, designated GST mu or GSTM1. This particular enzyme is of considerable toxicological interest because it appears to be involved in the subsequent detoxification of toxic metabolites produced from chemicals in cigarette smoke by the CYP1A1 enzyme. The identified polymorphism in this glutathione transferase gene involves a total absence of functional enzyme in as many as half of all Caucasians studied. This lack of a Phase II enzyme appears to be associated with increased susceptibility to lung cancer. By grouping individuals on the basis of both variant CYP1A1 genes and the deletion or presence of a functional GSTM1 gene, it has been demonstrated that the risk of developing smoking-induced lung cancer varies significantly (Kawajiri, Watanabe and Hayashi 1994). In particular, individuals displaying one rare CYP1A1 gene alteration, in combination with an absence of the GSTM1 gene, were at higher risk (as much as ninefold) of developing lung cancer when exposed to a relatively low level of cigarette smoke. Interestingly, there appear to be interethnic differences in the significance of variant genes which necessitate further study in order to elucidate the precise role of such alterations in susceptibility to disease (Kalow 1962; Nebert and McKinnon 1994; Kawajiri, Watanabe and Hayashi 1994).
Synergistic effect of two or more polymorphisms on the toxic response
A toxic response to an environmental agent may be greatly exaggerated by the combination of two pharmacogenetic defects in the same individual, for example, the combined effects of the N-acetyltransferase (NAT2) polymorphism and the glucose-6-phosphate dehydrogenase (G6PD) polymorphism.
Occupational exposure to arylamines constitutes a grave risk of urinary bladder cancer. Since the elegant studies of Cartwright in 1954, it has become clear that the N-acetylator status is a determinant of azo-dye-induced bladder cancer. There is a highly significant correlation between the slow-acetylator phenotype and the occurrence of bladder cancer, as well as the degree of invasiveness of this cancer in the bladder wall. On the contrary, there is a significant association between the rapid-acetylator phenotype and the incidence of colorectal carcinoma. The N-acetyltransferase (NAT1, NAT2) genes have been cloned and sequenced, and DNA-based assays are now able to detect the more than a dozen allelic variants which account for the slow-acetylator phenotype. The NAT2 gene is polymorphic and responsible for most of the variability in toxic response to environmental chemicals (Weber 1987; Grant 1993).
Glucose-6-phosphate dehydrogenase (G6PD) is an enzyme critical in the generation and maintenance of NADPH. Low or absent G6PD activity can lead to severe drug- or xenobiotic-induced haemolysis, due to the absence of normal levels of reduced glutathione (GSH) in the red blood cell. G6PD deficiency affects at least 300 million people worldwide. More than 10% of African-American males exhibit the less severe phenotype, while certain Sardinian communities exhibit the more severe “Mediterranean type” at frequencies as high as one in every three persons. The G6PD gene has been cloned and localized to the X chromosome, and numerous diverse point mutations account for the large degree of phenotypic heterogeneity seen in G6PD-deficient individuals (Beutler 1992).
Thiozalsulphone, an arylamine sulpha drug, was found to cause a bimodal distribution of haemolytic anaemia in the treated population. When treated with certain drugs, individuals with the combination of G6PD deficiency plus the slow-acetylator phenotype are more affected than those with the G6PD deficiency alone or the slow-acetylator phenotype alone. G6PD-deficient slow acetylators are at least 40 times more susceptible than normal-G6PD rapid acetylators to thiozalsulphone-induced haemolysis.
Effect of genetic polymorphisms on exposure assessment
Exposure assessment and biomonitoring (figure 1) also requires information on the genetic make-up of each individual. Given identical exposure to a hazardous chemical, the level of haemoglobin adducts (or other biomarkers) might vary by two or three orders of magnitude among individuals, depending upon each person’s metabolic fingerprint.
The same combined pharmacogenetics has been studied in chemical factory workers in Germany (table 1). Haemoglobin adducts among workers exposed to aniline and acetanilide are by far the highest in G6PD-deficient slow acetylators, as compared with the other possible combined pharmacogenetic phenotypes. This study has important implications for exposure assessment. These data demonstrate that, although two individuals might be exposed to the same ambient level of hazardous chemical in the work place, the amount of exposure (via biomarkers such as haemoglobin adducts) might be estimated to be two or more orders of magnitude less, due to the underlying genetic predisposition of the individual. Likewise, the resulting risk of an adverse health effect may vary by two or more orders of magnitude.
Table 1: Haemoglobin adducts in workers exposed to aniline and acetanilide
| Acetylator status | G6PD deficiency | |||
| Fast | Slow | No | Yes | Hgb adducts |
| + | + | 2 | ||
| + | + | 30 | ||
| + | + | 20 | ||
| + | + | 100 | ||
Source: Adapted from Lewalter and Korallus 1985.
Genetic differences in binding as well as metabolism
It should be emphasized that the same case made here for meta-bolism can also be made for binding. Heritable differences in the binding of environmental agents will greatly affect the toxic response. For example, differences in the mouse cdm gene can profoundly affect individual sensitivity to cadmium-induced testicular necrosis (Taylor, Heiniger and Meier 1973). Differences in the binding affinity of the Ah receptor are likely affect dioxin-induced toxicity and cancer (Nebert, Petersen and Puga 1991; Nebert, Puga and Vasiliou 1993).
Figure 5 summarizes the role of metabolism and binding in toxicity and cancer. Toxic agents, as they exist in the environment or following metabolism or binding, elicit their effects by either a genotoxic pathway (in which damage to DNA occurs) or a non-genotoxic pathway (in which DNA damage and mutagenesis need not occur). Interestingly, it has recently become clear that “classical” DNA-damaging agents can operate via a reduced glutathione (GSH)-dependent nongenotoxic signal transduction pathway, which is initiated on or near the cell surface in the absence of DNA and outside the cell nucleus (Devary et al. 1993). Genetic differences in metabolism and binding remain, however, as the major determinants in controlling different individual toxic responses.
Figure 5. The general means by which toxicity occurs

Role of Drug-metabolizing Enzymesin Cellular Function
Genetically based variation in drug-metabolizing enzyme function is of major importance in determining individual response to chemicals. These enzymes are pivotal in determining the fate and time course of a foreign chemical following exposure.
As illustrated in figure 5, the importance of drug-metabolizing enzymes in individual susceptibility to chemical exposure may in fact present a far more complex issue than is evident from this simple discussion of xenobiotic metabolism. In other words, during the past two decades, genotoxic mechanisms (measurements of DNA adducts and protein adducts) have been greatly emphasized. However, what if nongenotoxic mechanisms are at least as important as genotoxic mechanisms in causing toxic responses?
As mentioned earlier, the physiological roles of many drug-metabolizing enzymes involved in xenobiotic metabolism have not been accurately defined. Nebert (1994) has proposed that, because of their presence on this planet for more than 3.5 billion years, drug-metabolizing enzymes were originally (and are now still primarily) responsible for regulating the cellular levels of many nonpeptide ligands important in the transcriptional activation of genes affecting growth, differentiation, apoptosis, homeostasis and neuroendocrine functions. Furthermore, the toxicity of most, if not all, environmental agents occurs by means of agonist or antagonist action on these signal transduction pathways (Nebert 1994). Based on this hypothesis, genetic variability in drug-metabolizing enzymes may have quite dramatic effects on many critical biochemical processes within the cell, thereby leading to important differences in toxic response. It is indeed possible that such a scenario may also underlie many idiosyncratic adverse reactions encountered in patients using commonly prescribed drugs.
Conclusions
The past decade has seen remarkable progress in our understanding of the genetic basis of differential response to chemicals in drugs, foods and environmental pollutants. Drug-metabolizing enzymes have a profound influence on the way humans respond to chemicals. As our awareness of drug-metabolizing enzyme multiplicity continues to evolve, we are increasingly able to make improved assessments of toxic risk for many drugs and environmental chemicals. This is perhaps most clearly illustrated in the case of the CYP2D6 cytochrome P450 enzyme. Using relatively simple DNA-based tests, it is possible to predict the likely response of any drug predominantly metabolized by this enzyme; this prediction will ensure the safer use of valuable, yet potentially toxic, medication.
The future will no doubt see an explosion in the identification of further polymorphisms (phenotypes) involving drug-metabolizing enzymes. This information will be accompanied by improved, minimally invasive DNA-based tests to identify genotypes in human populations.
Such studies should be particularly informative in evaluating the role of chemicals in the many environmental diseases of presently unknown origin. The consideration of multiple drug-metabolizing enzyme polymorphisms, in combination (e.g., table 1), is also likely to represent a particularly fertile research area. Such studies will clarify the role of chemicals in the causation of cancers. Collectively, this information should enable the formulation of increasingly individualized advice on avoidance of chemicals likely to be of individual concern. This is the field of preventive toxicology. Such advice will no doubt greatly assist all individuals in coping with the ever increasing chemical burden to which we are exposed.